

Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin
- PMID: 33514319
- PMCID: PMC7817968
- DOI: 10.1186/s12862-020-01732-2
Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin
Abstract
Background: The SARS-CoV-2 pandemic is one of the greatest global medical and social challenges that have emerged in recent history. Human coronavirus strains discovered during previous SARS outbreaks have been hypothesized to pass from bats to humans using intermediate hosts, e.g. civets for SARS-CoV and camels for MERS-CoV. The discovery of an intermediate host of SARS-CoV-2 and the identification of specific mechanism of its emergence in humans are topics of primary evolutionary importance. In this study we investigate the evolutionary patterns of 11 main genes of SARS-CoV-2. Previous studies suggested that the genome of SARS-CoV-2 is highly similar to the horseshoe bat coronavirus RaTG13 for most of the genes and to some Malayan pangolin coronavirus (CoV) strains for the receptor binding (RB) domain of the spike protein.
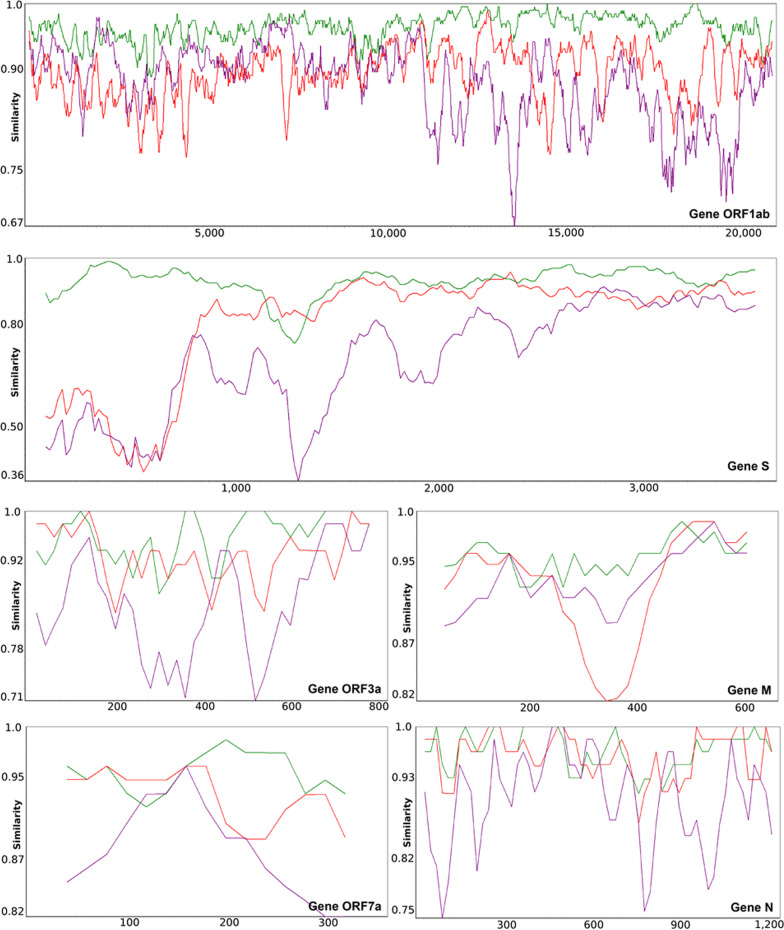
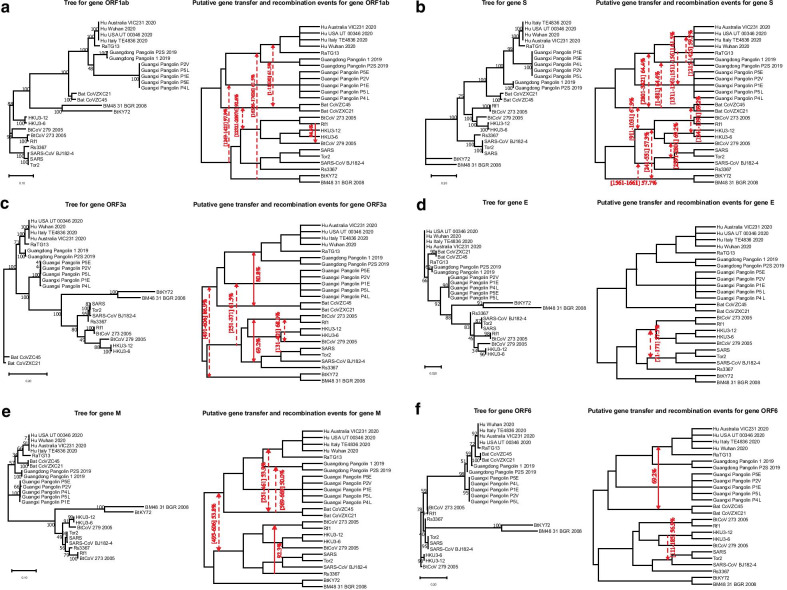
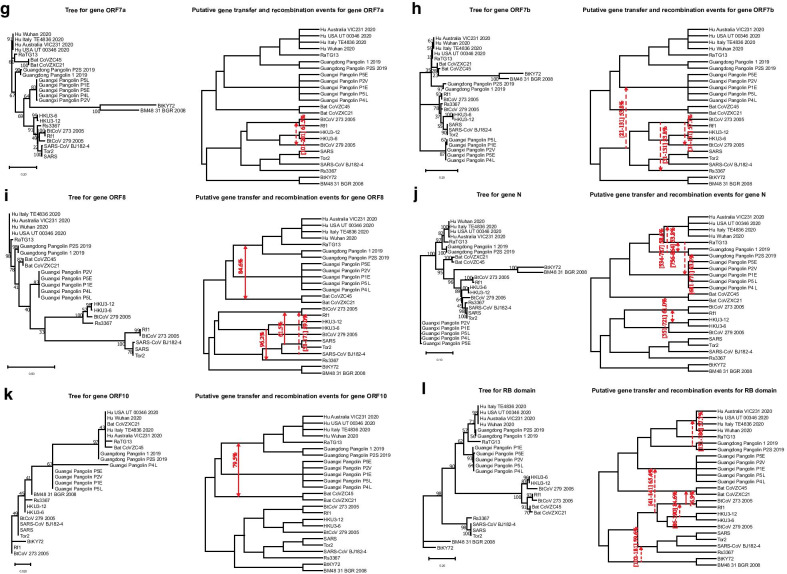
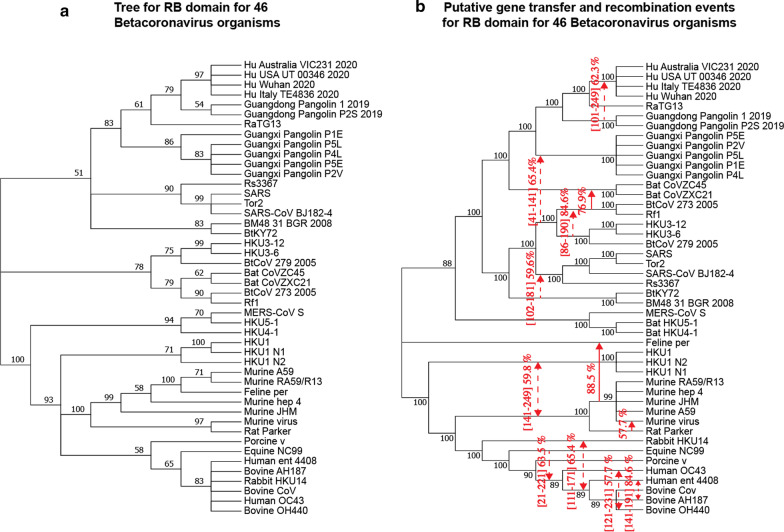
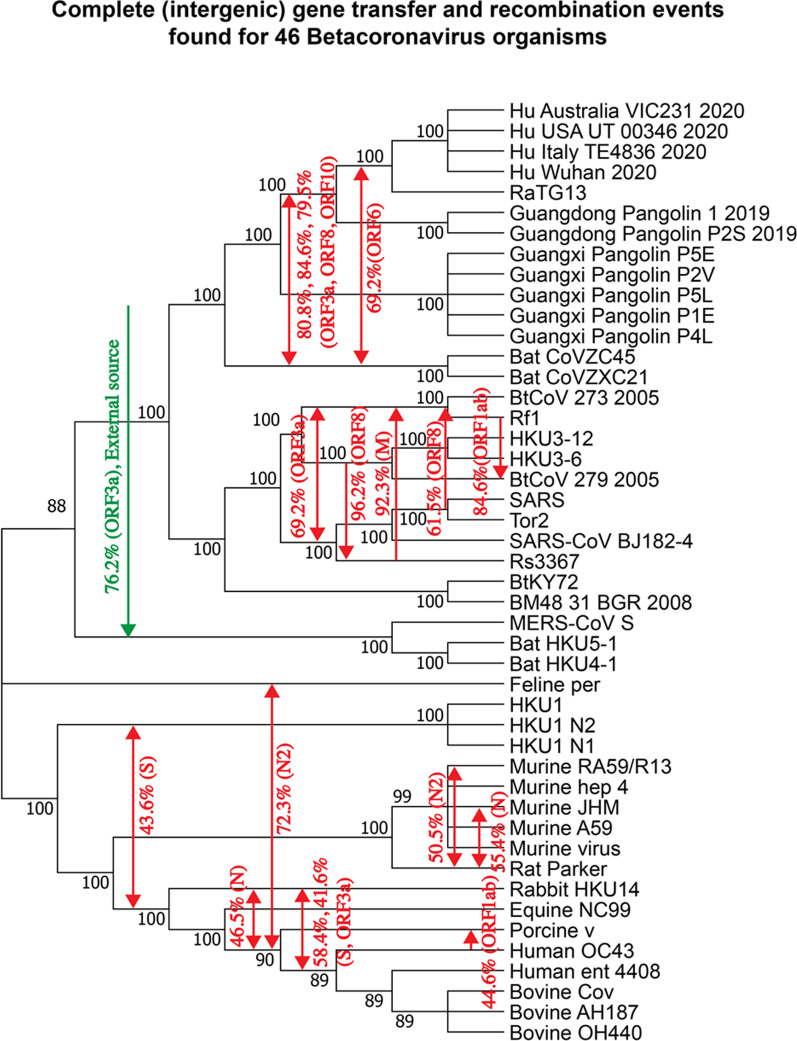
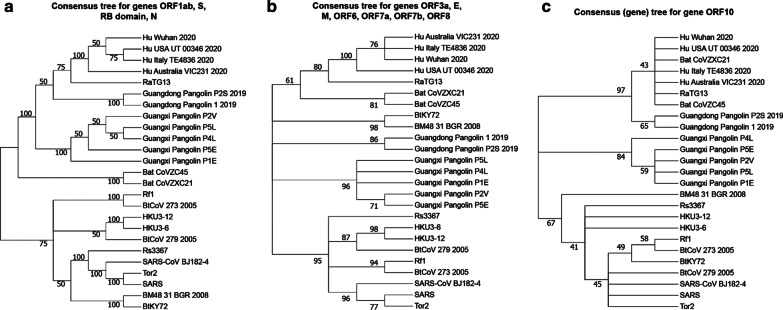
Results: We provide a detailed list of statistically significant horizontal gene transfer and recombination events (both intergenic and intragenic) inferred for each of 11 main genes of the SARS-CoV-2 genome. Our analysis reveals that two continuous regions of genes S and N of SARS-CoV-2 may result from intragenic recombination between RaTG13 and Guangdong (GD) Pangolin CoVs. Statistically significant gene transfer-recombination events between RaTG13 and GD Pangolin CoV have been identified in region [1215-1425] of gene S and region [534-727] of gene N. Moreover, some statistically significant recombination events between the ancestors of SARS-CoV-2, RaTG13, GD Pangolin CoV and bat CoV ZC45-ZXC21 coronaviruses have been identified in genes ORF1ab, S, ORF3a, ORF7a, ORF8 and N. Furthermore, topology-based clustering of gene trees inferred for 25 CoV organisms revealed a three-way evolution of coronavirus genes, with gene phylogenies of ORF1ab, S and N forming the first cluster, gene phylogenies of ORF3a, E, M, ORF6, ORF7a, ORF7b and ORF8 forming the second cluster, and phylogeny of gene ORF10 forming the third cluster.
Conclusions: The results of our horizontal gene transfer and recombination analysis suggest that SARS-CoV-2 could not only be a chimera virus resulting from recombination of the bat RaTG13 and Guangdong pangolin coronaviruses but also a close relative of the bat CoV ZC45 and ZXC21 strains. They also indicate that a GD pangolin may be an intermediate host of this dangerous virus.
Keywords: Consensus tree; Evolution of SARS-CoV-2; Gene evolution; Horizontal gene transfer; Phylogenetic network; Recombination.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous