

H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions
- PMID: 33514712
- PMCID: PMC7846766
- DOI: 10.1038/s41467-021-20940-y
H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions
Abstract
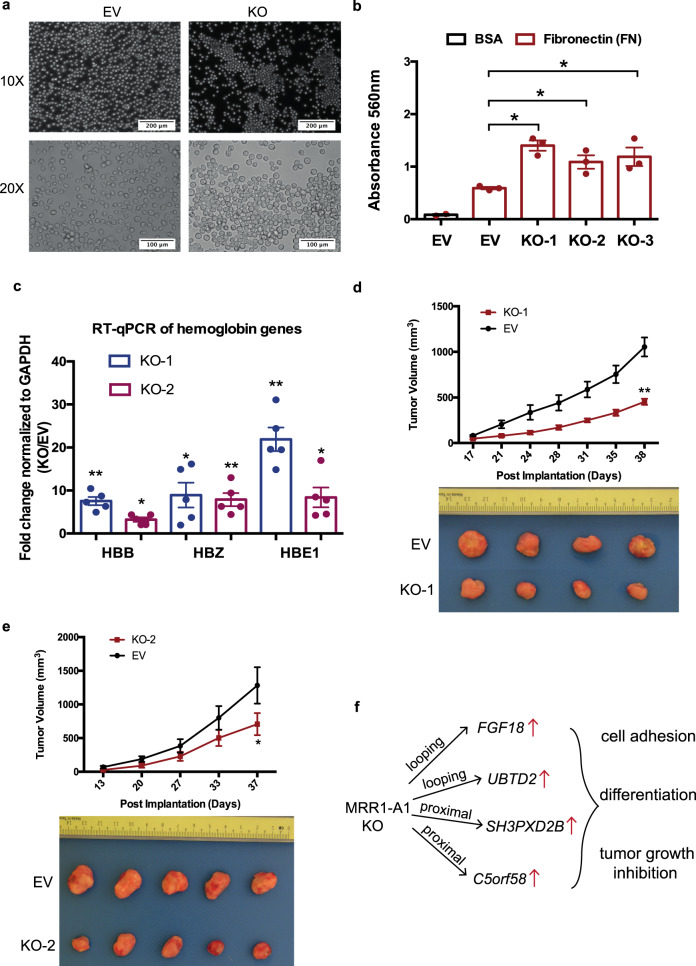
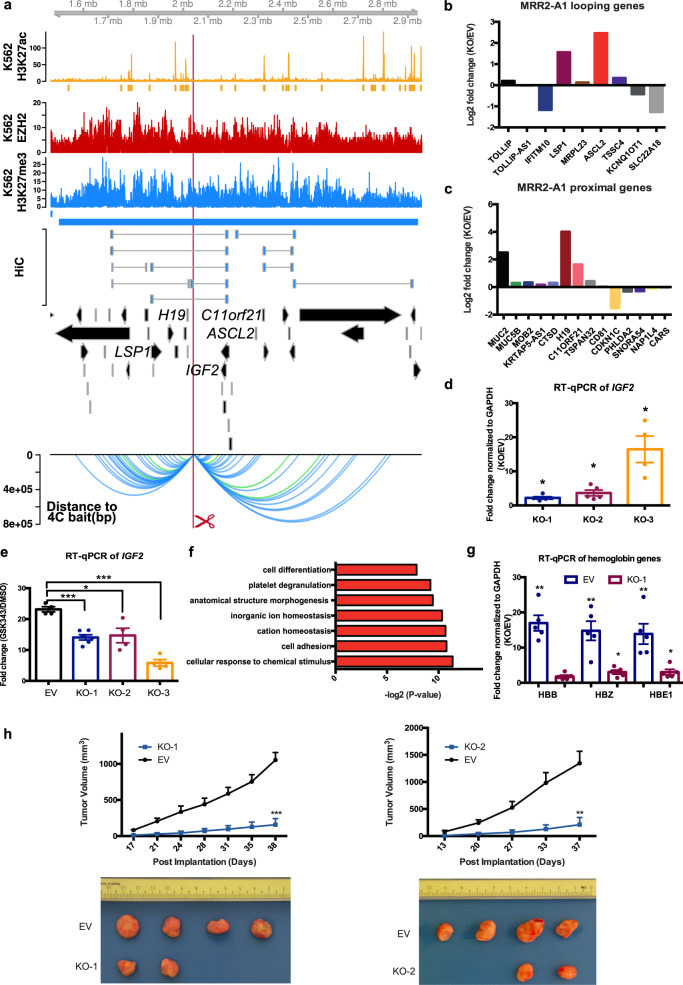
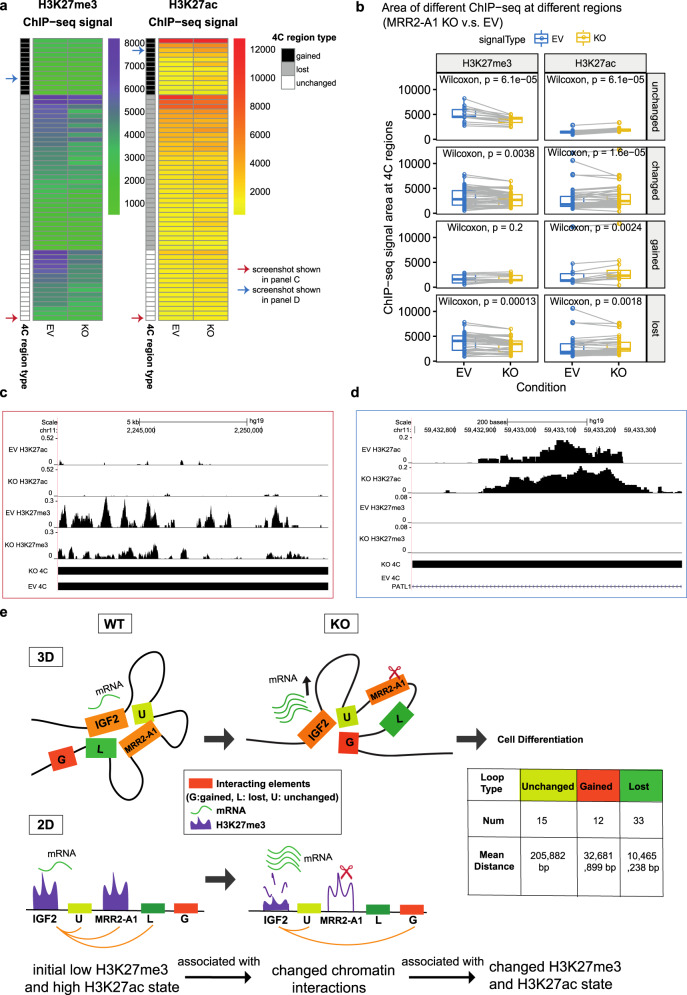
The mechanisms underlying gene repression and silencers are poorly understood. Here we investigate the hypothesis that H3K27me3-rich regions of the genome, defined from clusters of H3K27me3 peaks, may be used to identify silencers that can regulate gene expression via proximity or looping. We find that H3K27me3-rich regions are associated with chromatin interactions and interact preferentially with each other. H3K27me3-rich regions component removal at interaction anchors by CRISPR leads to upregulation of interacting target genes, altered H3K27me3 and H3K27ac levels at interacting regions, and altered chromatin interactions. Chromatin interactions did not change at regions with high H3K27me3, but regions with low H3K27me3 and high H3K27ac levels showed changes in chromatin interactions. Cells with H3K27me3-rich regions knockout also show changes in phenotype associated with cell identity, and altered xenograft tumor growth. Finally, we observe that H3K27me3-rich regions-associated genes and long-range chromatin interactions are susceptible to H3K27me3 depletion. Our results characterize H3K27me3-rich regions and their mechanisms of functioning via looping.
Conflict of interest statement
The authors declare no competing interests.
Figures







References
-
- Akincilar SC, et al. Long-range chromatin interactions drive mutant TERT promoter activation. Cancer Discov. 2016;6:1276–1291. doi: 10.1158/2159-8290.CD-16-0177. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
