

Splicing-associated chromatin signatures: a combinatorial and position-dependent role for histone marks in splicing definition
- PMID: 33514745
- PMCID: PMC7846797
- DOI: 10.1038/s41467-021-20979-x
Splicing-associated chromatin signatures: a combinatorial and position-dependent role for histone marks in splicing definition
Abstract
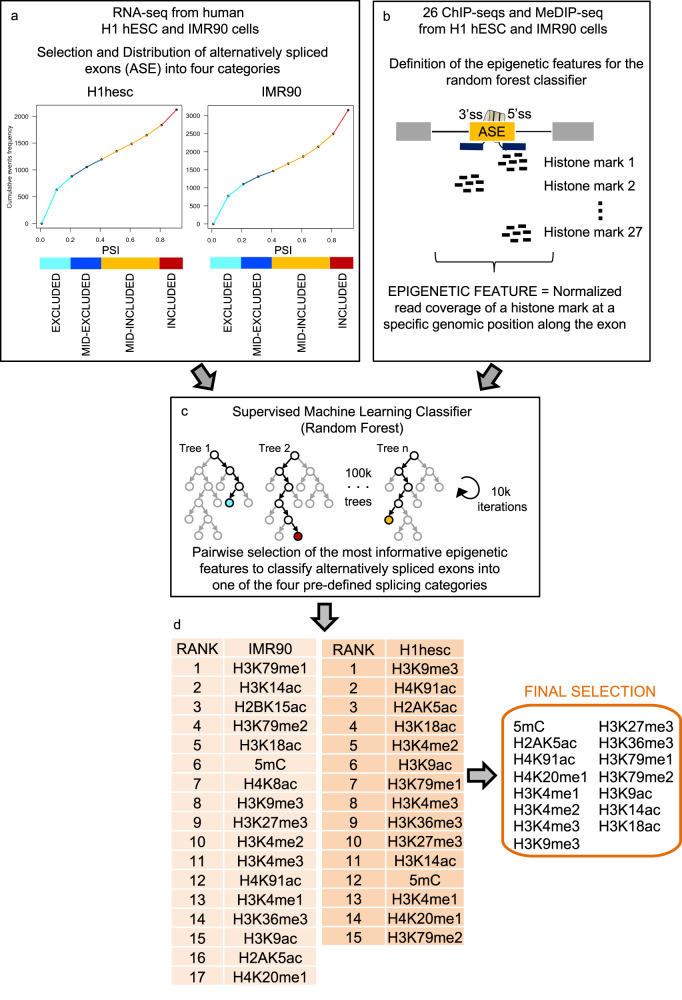
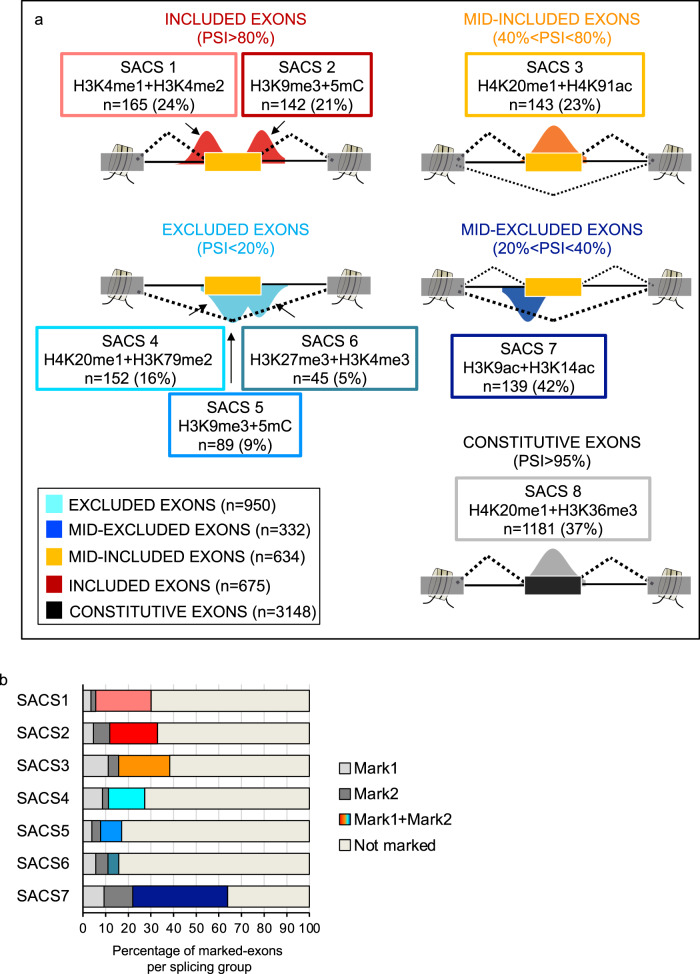
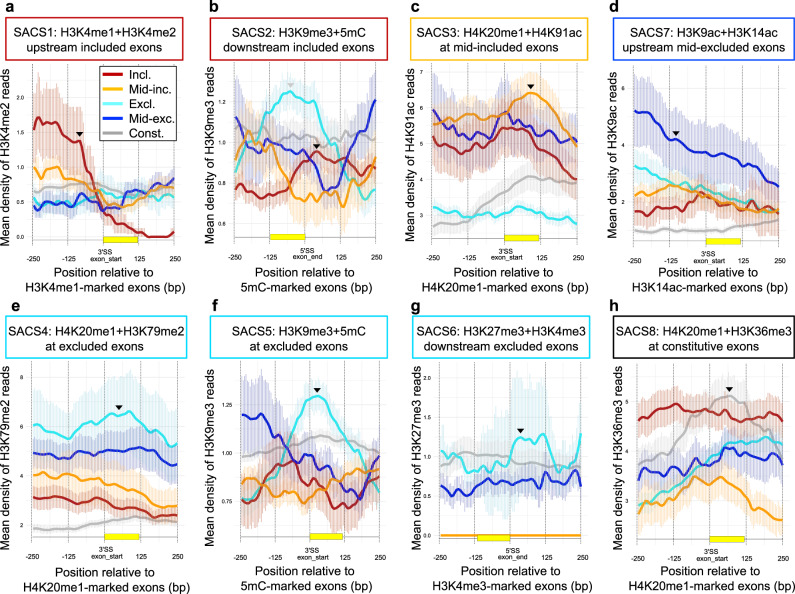
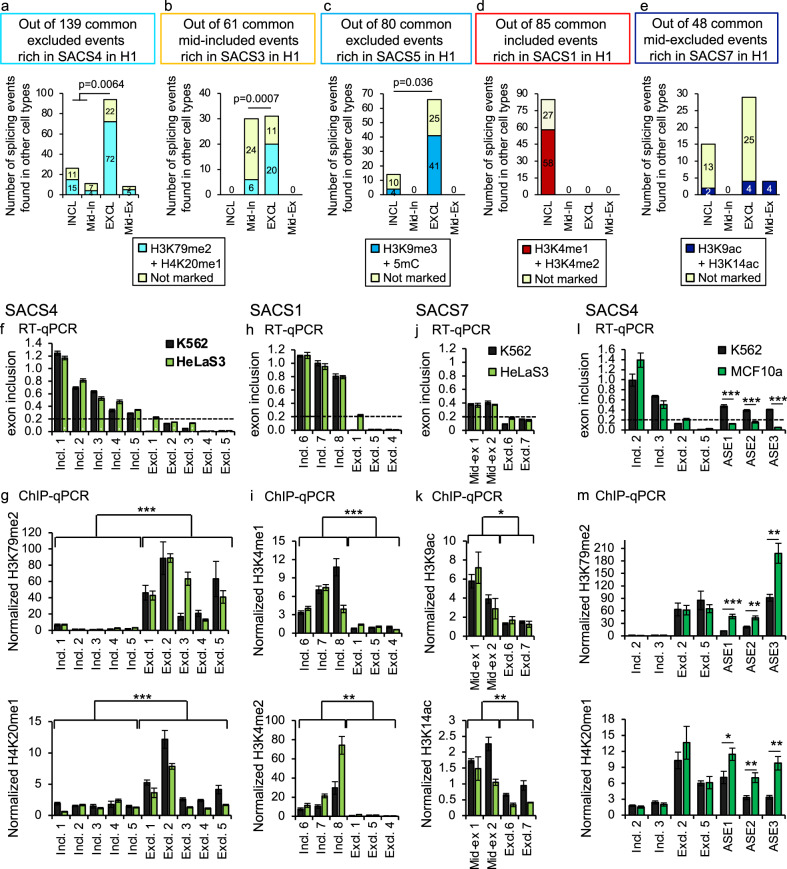
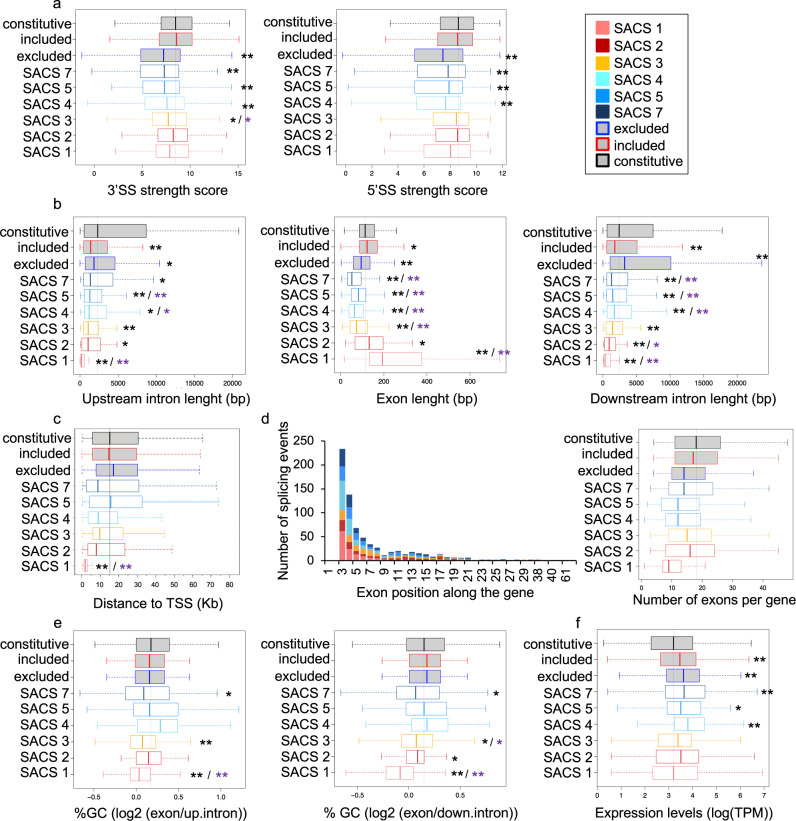
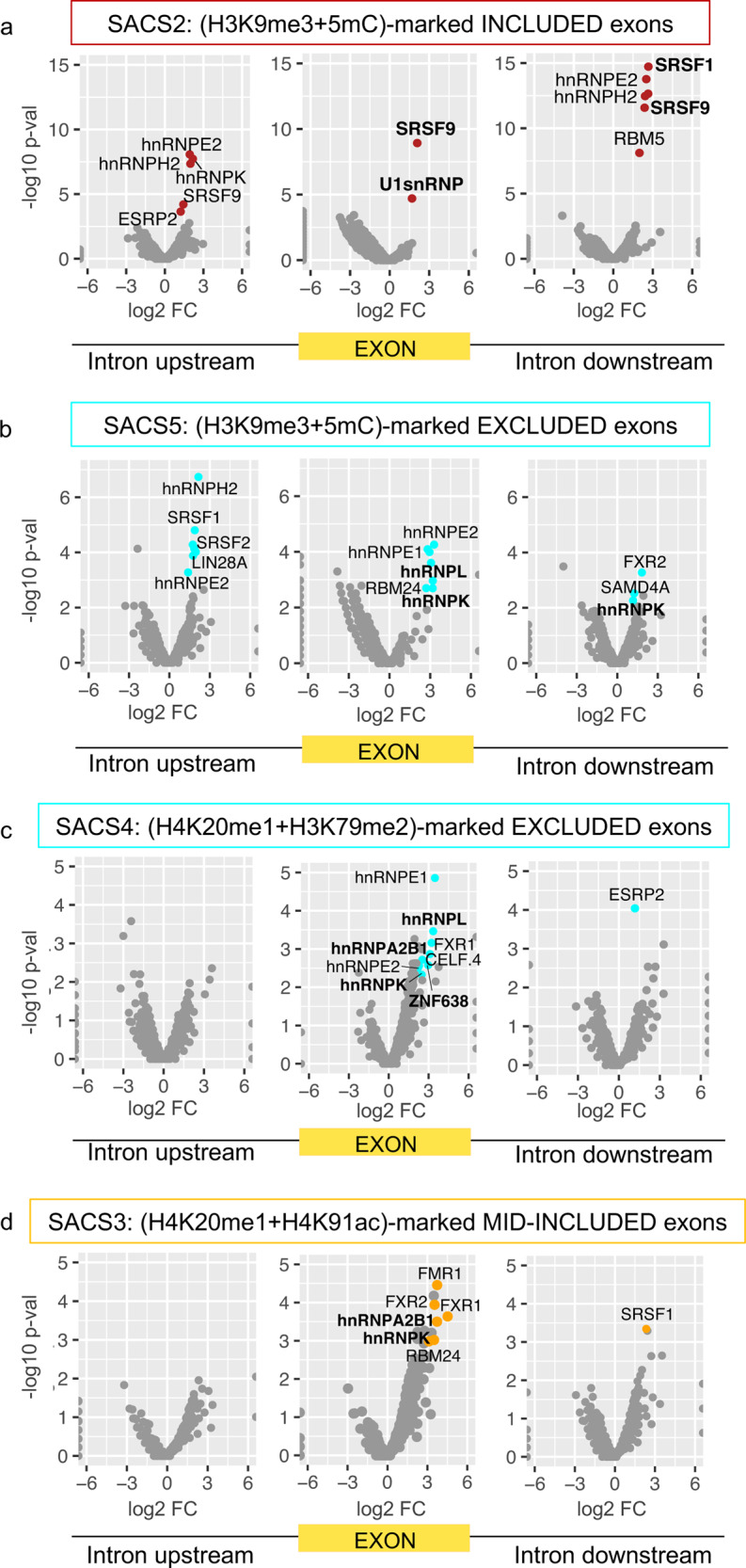
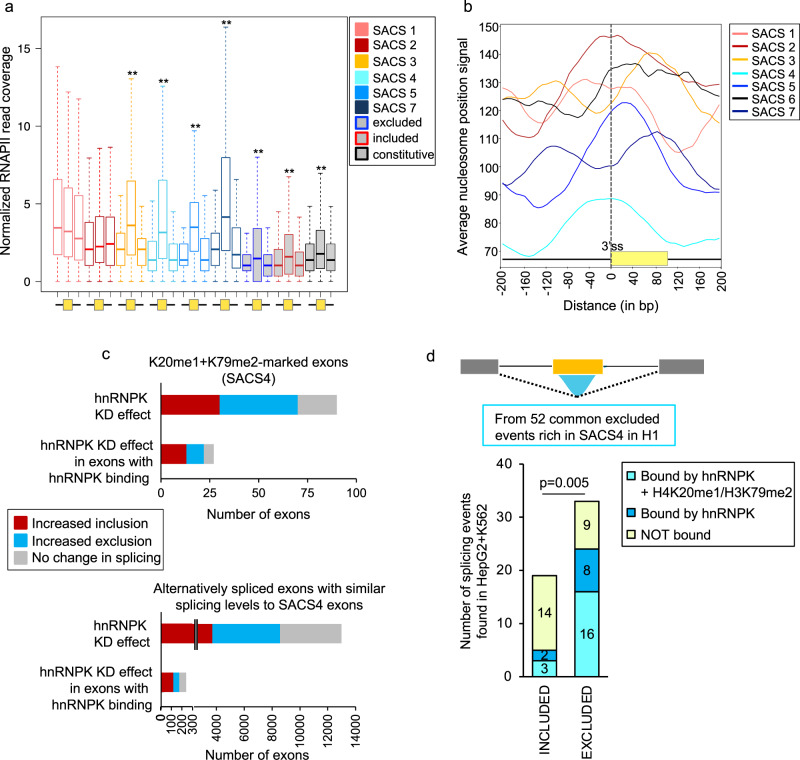
Alternative splicing relies on the combinatorial recruitment of splicing regulators to specific RNA binding sites. Chromatin has been shown to impact this recruitment. However, a limited number of histone marks have been studied at a global level. In this work, a machine learning approach, applied to extensive epigenomics datasets in human H1 embryonic stem cells and IMR90 foetal fibroblasts, has identified eleven chromatin modifications that differentially mark alternatively spliced exons depending on the level of exon inclusion. These marks act in a combinatorial and position-dependent way, creating characteristic splicing-associated chromatin signatures (SACS). In support of a functional role for SACS in coordinating splicing regulation, changes in the alternative splicing of SACS-marked exons between ten different cell lines correlate with changes in SACS enrichment levels and recruitment of the splicing regulators predicted by RNA motif search analysis. We propose the dynamic nature of chromatin modifications as a mechanism to rapidly fine-tune alternative splicing when necessary.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
