

McSplicer: a probabilistic model for estimating splice site usage from RNA-seq data
- PMID: 33515239
- PMCID: PMC8337008
- DOI: 10.1093/bioinformatics/btab050
McSplicer: a probabilistic model for estimating splice site usage from RNA-seq data
Abstract
Motivation: Alternative splicing removes intronic sequences from pre-mRNAs in alternative ways to produce different forms (isoforms) of mature mRNA. The composition of expressed transcripts gives specific functionalities to cells in a particular condition or developmental stage. In addition, a large fraction of human disease mutations affect splicing and lead to aberrant mRNA and protein products. Current methods that interrogate the transcriptome based on RNA-seq either suffer from short read length when trying to infer full-length transcripts, or are restricted to predefined units of alternative splicing that they quantify from local read evidence.
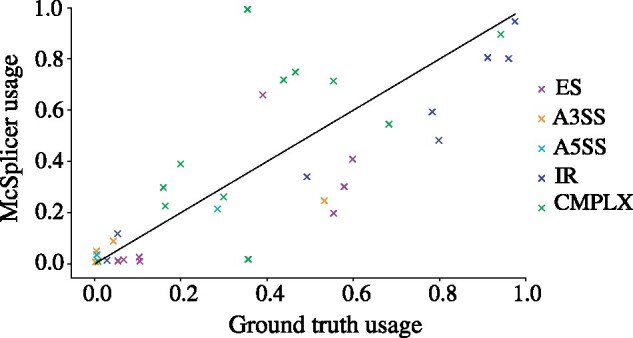
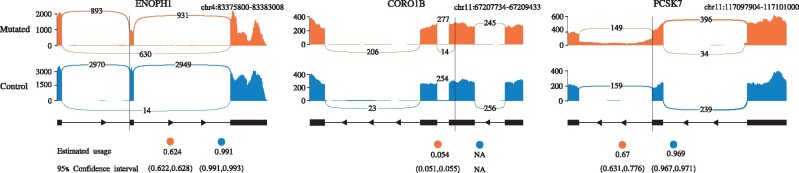
Results: Instead of attempting to quantify individual outcomes of the splicing process such as local splicing events or full-length transcripts, we propose to quantify alternative splicing using a simplified probabilistic model of the underlying splicing process. Our model is based on the usage of individual splice sites and can generate arbitrarily complex types of splicing patterns. In our implementation, McSplicer, we estimate the parameters of our model using all read data at once and we demonstrate in our experiments that this yields more accurate estimates compared to competing methods. Our model is able to describe multiple effects of splicing mutations using few, easy to interpret parameters, as we illustrate in an experiment on RNA-seq data from autism spectrum disorder patients.
Availability: McSplicer source code is available at https://github.com/canzarlab/McSplicer and has been deposited in archived format at https://doi.org/10.5281/zenodo.4449881.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press.
Figures

References
-
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525–527. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
