

STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway
- PMID: 33515644
- PMCID: PMC8573791
- DOI: 10.1016/j.jhep.2021.01.028
STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway
Abstract
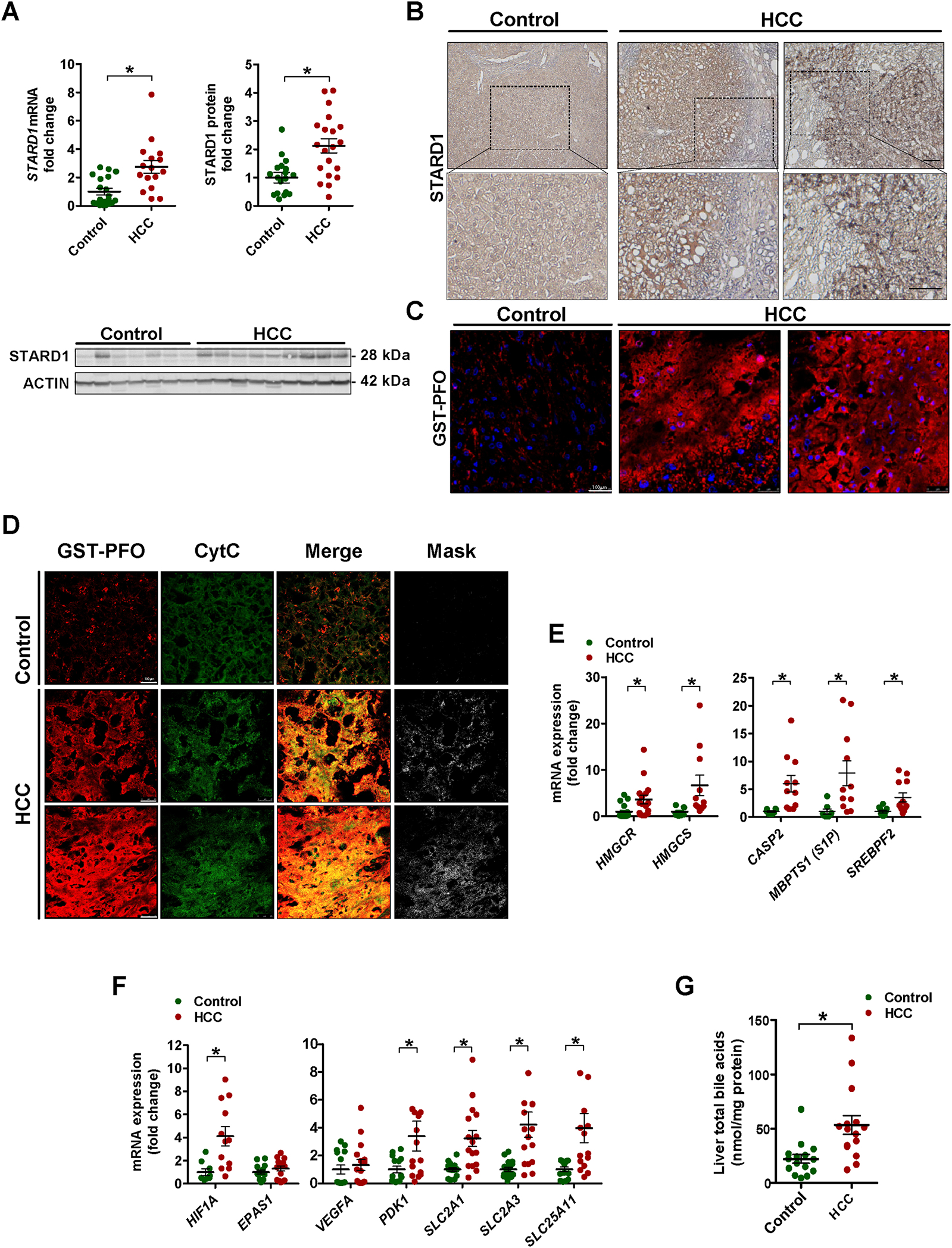
Background & aims: Besides their physiological role in bile formation and fat digestion, bile acids (BAs) synthesised from cholesterol in hepatocytes act as signalling molecules that modulate hepatocellular carcinoma (HCC). Trafficking of cholesterol to mitochondria through steroidogenic acute regulatory protein 1 (STARD1) is the rate-limiting step in the alternative pathway of BA generation, the physiological relevance of which is not well understood. Moreover, the specific contribution of the STARD1-dependent BA synthesis pathway to HCC has not been previously explored.
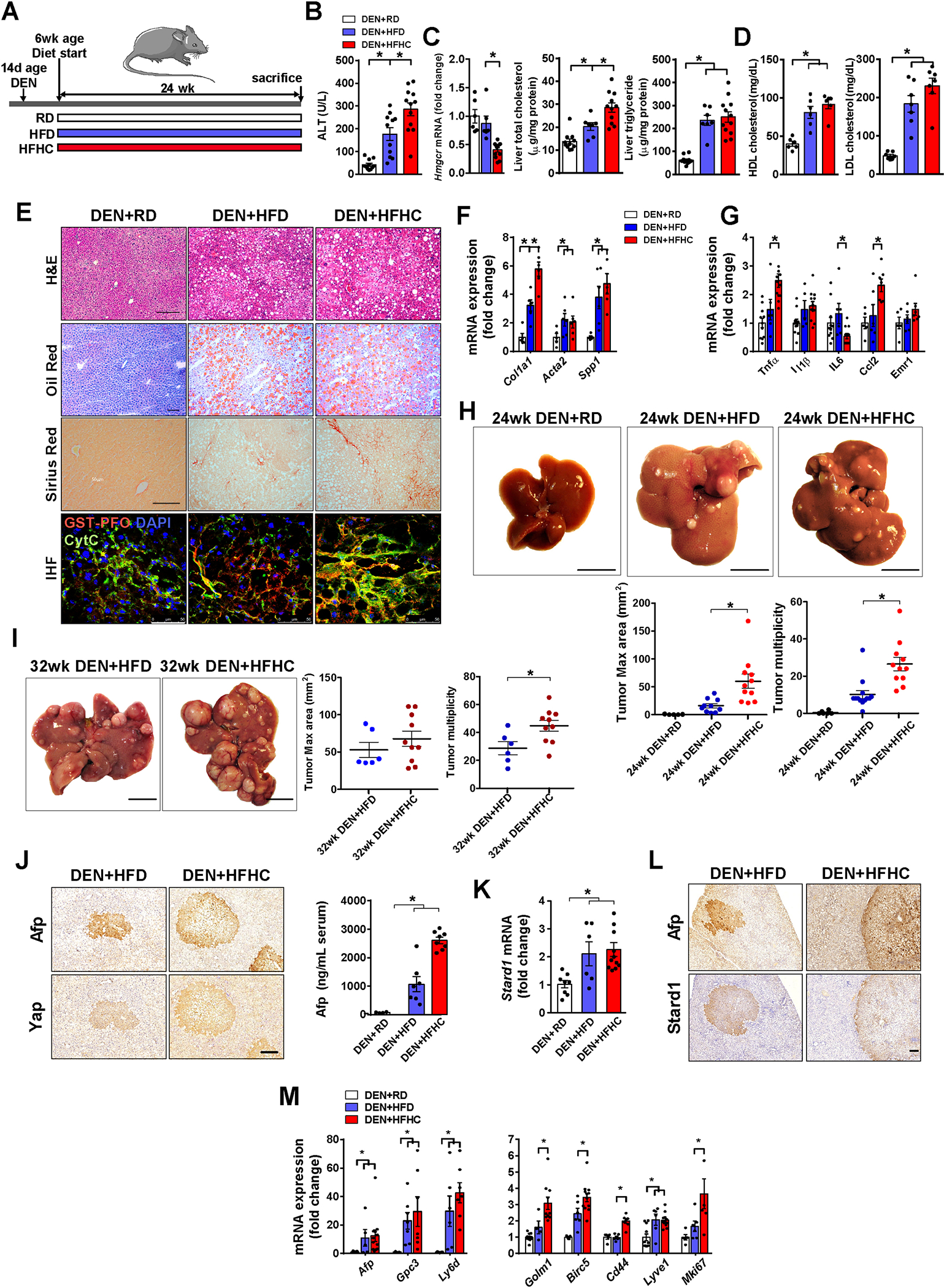
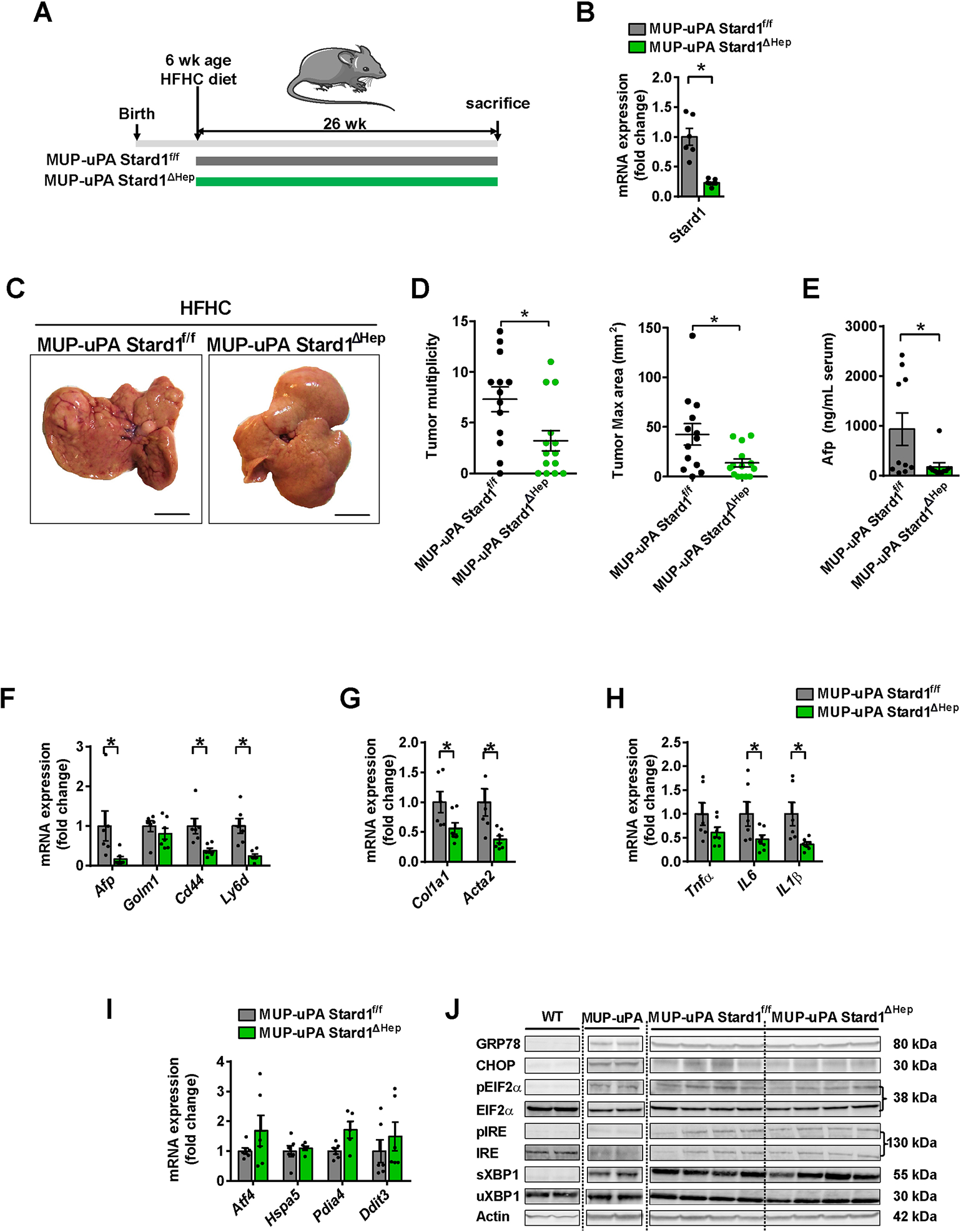
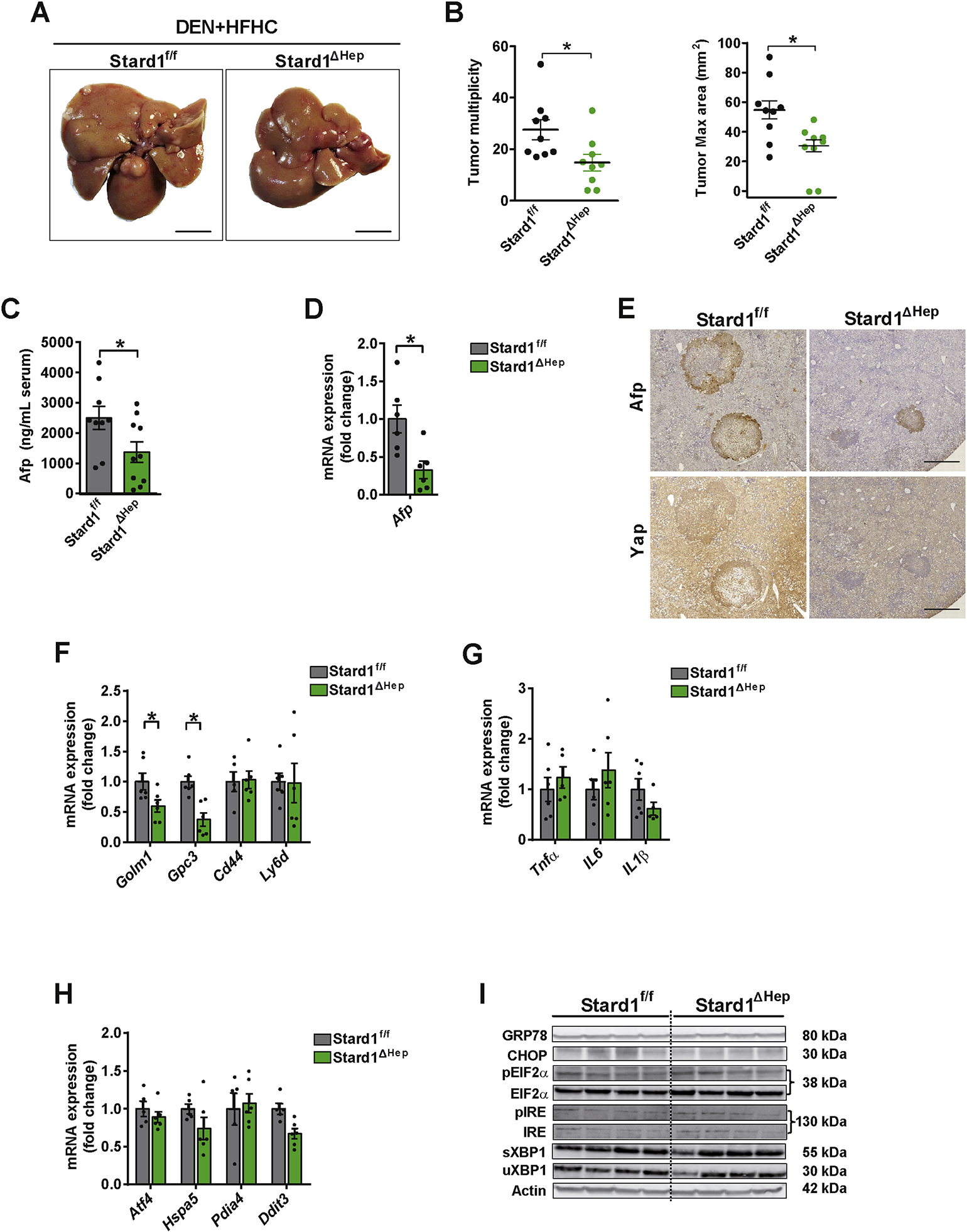
Methods: STARD1 expression was analyzed in a cohort of human non-alcoholic steatohepatitis (NASH)-derived HCC specimens. Experimental NASH-driven HCC models included MUP-uPA mice fed a high-fat high-cholesterol (HFHC) diet and diethylnitrosamine (DEN) treatment in wild-type (WT) mice fed a HFHC diet. Molecular species of BAs and oxysterols were analyzed by mass spectrometry. Effects of NASH-derived BA profiles were investigated in tumour-initiated stem-like cells (TICs) and primary mouse hepatocytes (PMHs).
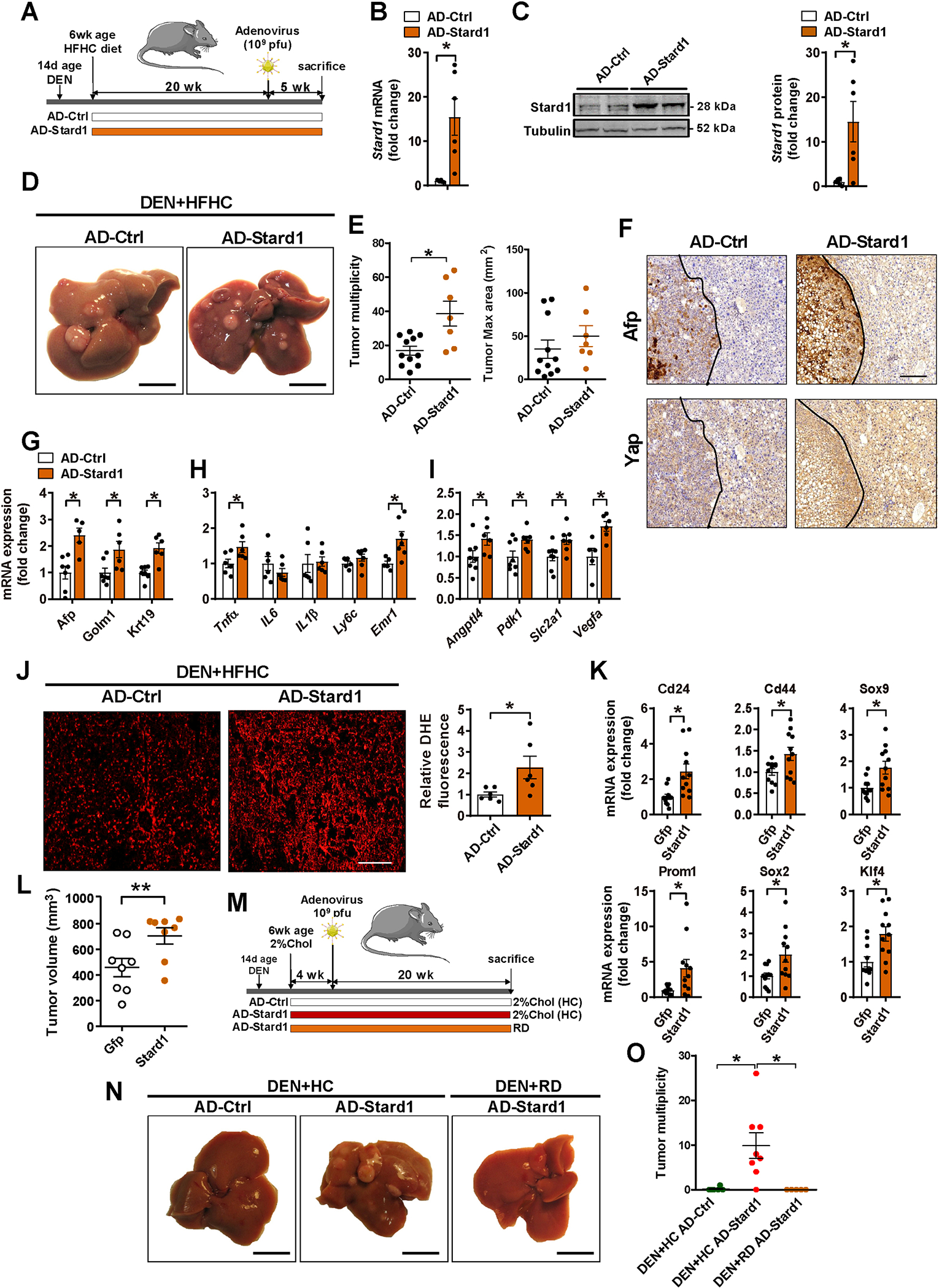
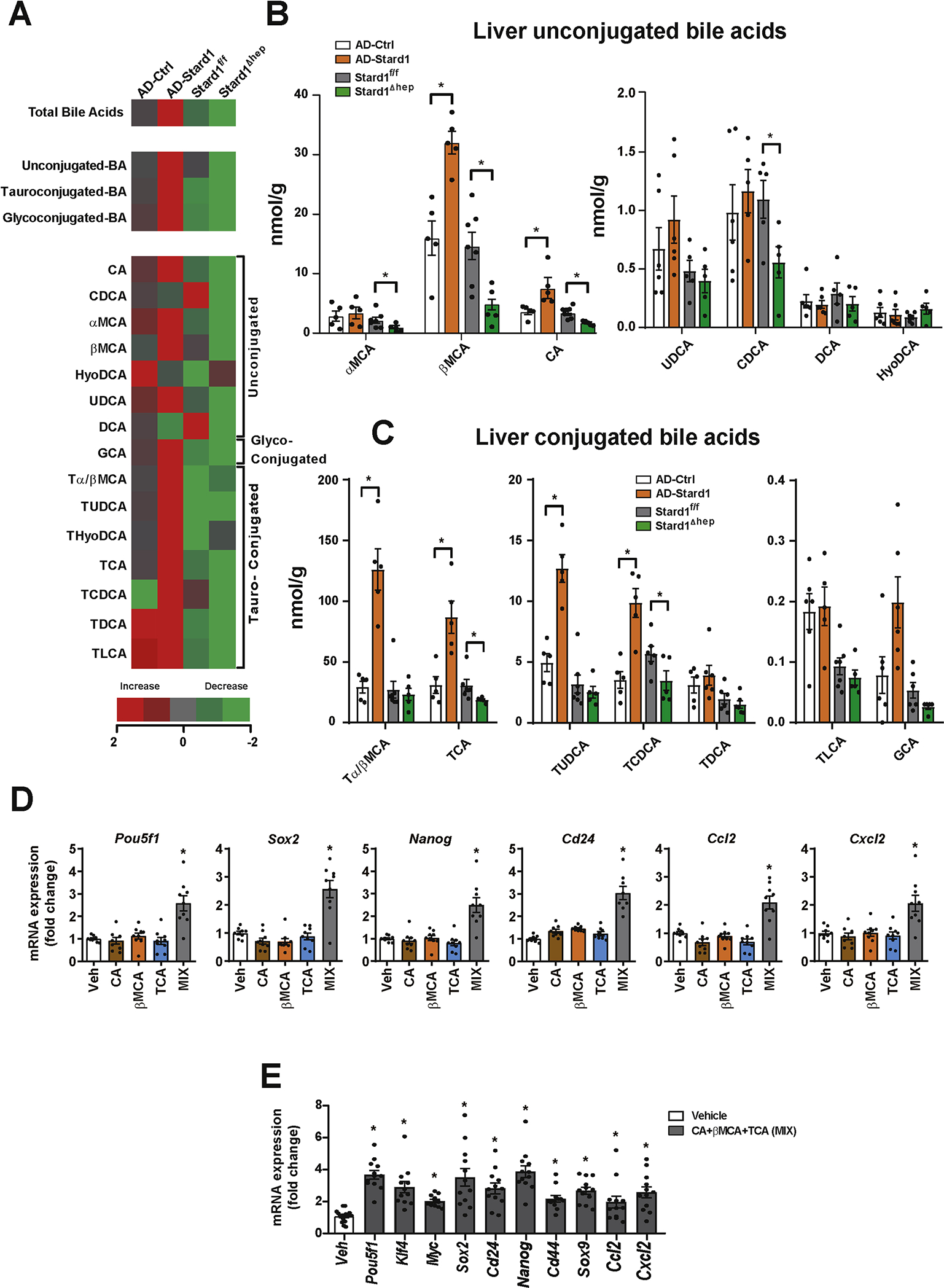
Results: Patients with NASH-associated HCC exhibited increased hepatic expression of STARD1 and an enhanced BA pool. Using NASH-driven HCC models, STARD1 overexpression in WT mice increased liver tumour multiplicity, whereas hepatocyte-specific STARD1 deletion (Stard1ΔHep) in WT or MUP-uPA mice reduced tumour burden. These findings mirrored the levels of unconjugated primary BAs, β-muricholic acid and cholic acid, and their tauroconjugates in STARD1-overexpressing and Stard1ΔHep mice. Incubation of TICs or PMHs with a mix of BAs mimicking this profile stimulated expression of genes involved in pluripotency, stemness and inflammation.
Conclusions: The study reveals a previously unrecognised role of STARD1 in HCC pathogenesis, wherein it promotes the synthesis of primary BAs through the mitochondrial pathway, the products of which act in TICs to stimulate self-renewal, stemness and inflammation.
Lay summary: Effective therapy for hepatocellular carcinoma (HCC) is limited because of our incomplete understanding of its pathogenesis. The contribution of the alternative pathway of bile acid (BA) synthesis to HCC development is unknown. We uncover a key role for steroidogenic acute regulatory protein 1 (STARD1) in non-alcoholic steatohepatitis-driven HCC, wherein it stimulates the generation of BAs in the mitochondrial acidic pathway, the products of which stimulate hepatocyte pluripotency and self-renewal, as well as inflammation.
Keywords: Bile acids; Cholesterol; Hepatocellular carcinoma; Mitochondria; Oxysterols; STARD1.
Copyright © 2021 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Conflicts of interest The authors declare no conflicts of interest that pertain to this work. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures
Comment in
-
Controlling cholesterol entry into mitochondria, a key step for hepatocarcinogenesis in non-alcoholic steatohepatitis-related hepatocellular carcinoma.Hepatobiliary Surg Nutr. 2021 Dec;10(6):890-892. doi: 10.21037/hbsn-21-406. Hepatobiliary Surg Nutr. 2021. PMID: 35004964 Free PMC article. No abstract available.
-
STARD1: a new rising StAR in cholesterol-mediated hepatocarcinogenesis.Hepatobiliary Surg Nutr. 2021 Dec;10(6):910-912. doi: 10.21037/hbsn-21-374. Hepatobiliary Surg Nutr. 2021. PMID: 35004970 Free PMC article. No abstract available.
References
-
- Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. The lancet oncology 2002;3:565–574. - PubMed
-
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England journal of medicine 2003;348:1625–1638. - PubMed
-
- Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391:1301–1314. - PubMed
-
- Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016;150:835–853. - PubMed
-
- Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nature reviews Gastroenterology & hepatology 2013;10:34–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
