

Genotype & phenotype in Lowe Syndrome: specific OCRL1 patient mutations differentially impact cellular phenotypes
- PMID: 33517444
- PMCID: PMC8091038
- DOI: 10.1093/hmg/ddab025
Genotype & phenotype in Lowe Syndrome: specific OCRL1 patient mutations differentially impact cellular phenotypes
Abstract
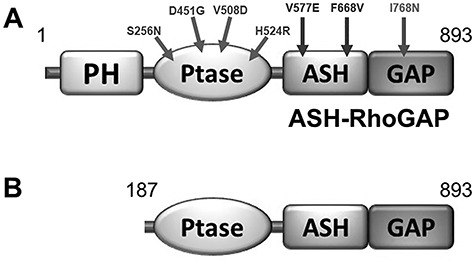
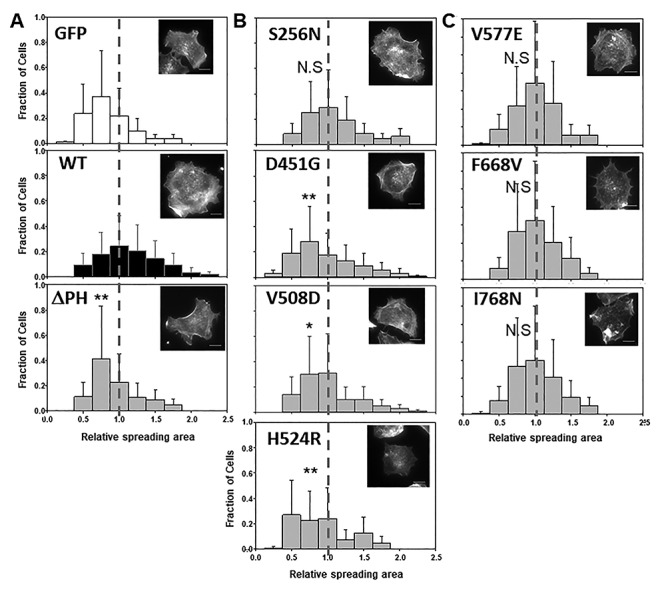
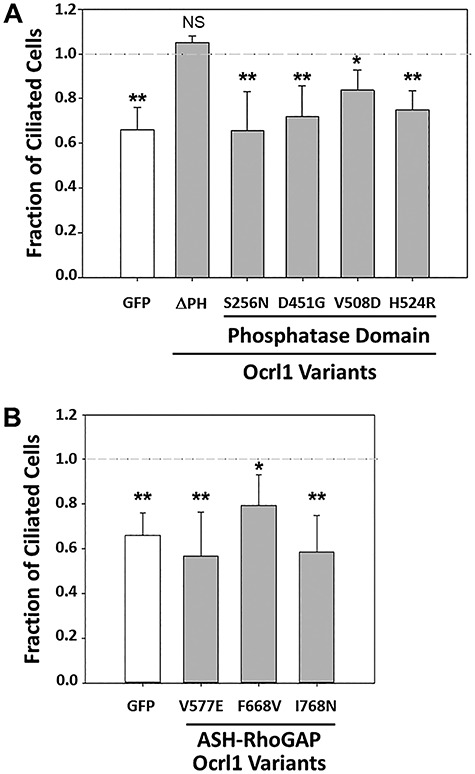
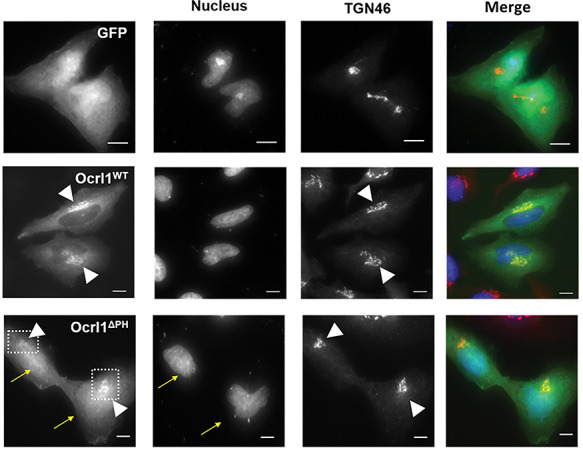
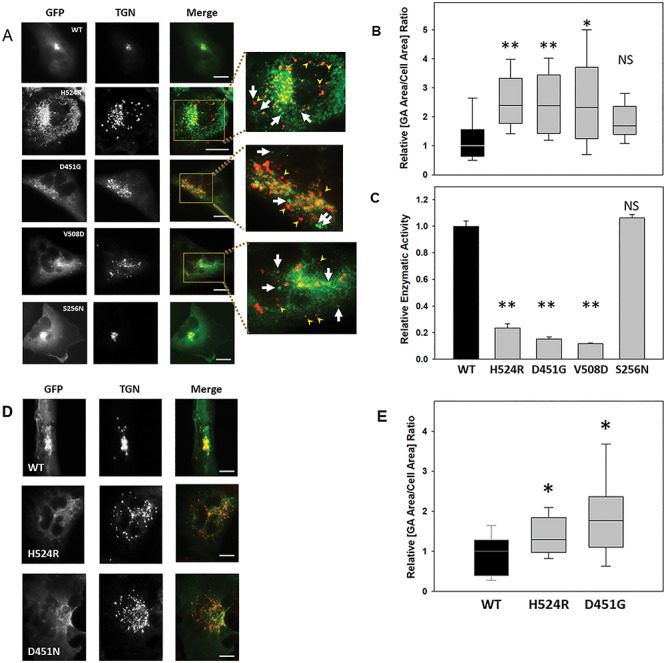
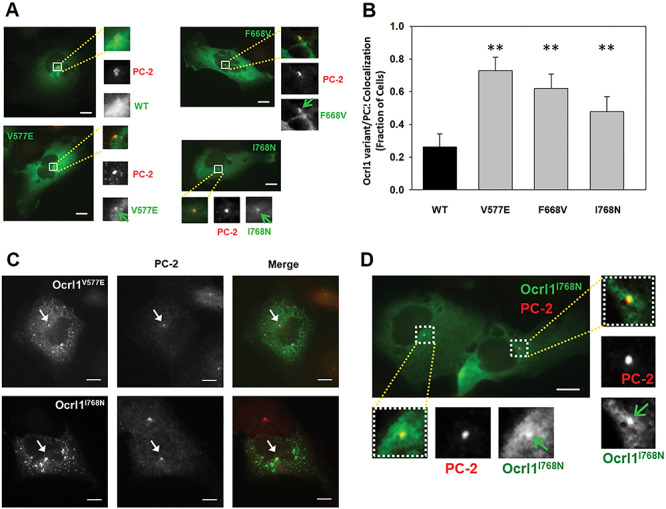
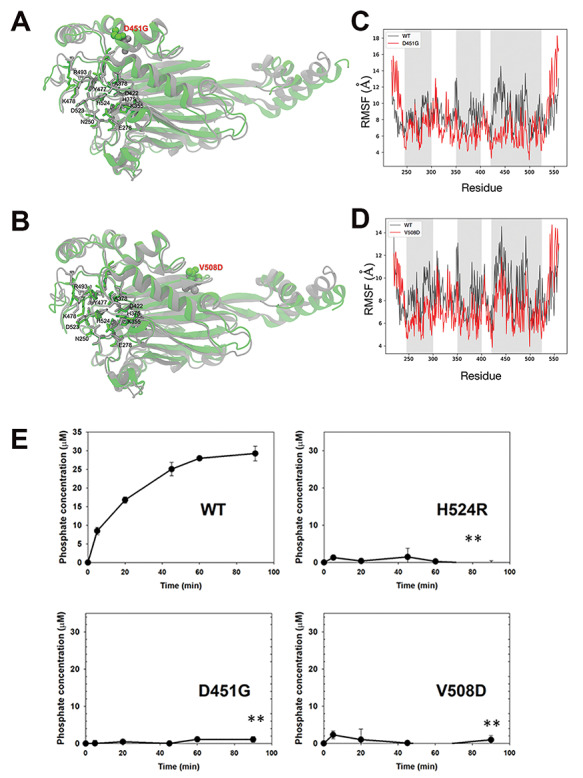
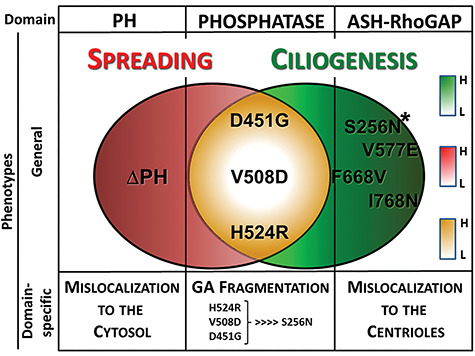
Lowe Syndrome (LS) is a lethal genetic disorder caused by mutations in the OCRL1 gene which encodes the lipid 5' phosphatase Ocrl1. Patients exhibit a characteristic triad of symptoms including eye, brain and kidney abnormalities with renal failure as the most common cause of premature death. Over 200 OCRL1 mutations have been identified in LS, but their specific impact on cellular processes is unknown. Despite observations of heterogeneity in patient symptom severity, there is little understanding of the correlation between genotype and its impact on phenotype. Here, we show that different mutations had diverse effects on protein localization and on triggering LS cellular phenotypes. In addition, some mutations affecting specific domains imparted unique characteristics to the resulting mutated protein. We also propose that certain mutations conformationally affect the 5'-phosphatase domain of the protein, resulting in loss of enzymatic activity and causing common and specific phenotypes (a conformational disease scenario). This study is the first to show the differential effect of patient 5'-phosphatase mutations on cellular phenotypes and introduces a conformational disease component in LS. This work provides a framework that explains symptom heterogeneity and can help stratify patients as well as to produce a more accurate prognosis depending on the nature and location of the mutation within the OCRL1 gene.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Attree, O., Olivos, I.M., Okabe, I., Bailey, L.C., Nelson, D.L., Lewis, R.A., McInnes, R.R. and Nussbaum, R.L. (1992) The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature, 358, 239–242. - PubMed
-
- Lowe, C.U., Terrey, M. and MacLachlan, E.A. (1952) Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. A.M.A. Am. J. Dis. Child., 83, 164–184. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
