

Diabetic Nephropathy: Novel Molecular Mechanisms and Therapeutic Targets
- PMID: 33519447
- PMCID: PMC7845653
- DOI: 10.3389/fphar.2020.586892
Diabetic Nephropathy: Novel Molecular Mechanisms and Therapeutic Targets
Abstract
Diabetic nephropathy (DN) is one of the major microvascular complications of diabetes mellitus and the leading cause of end-stage kidney disease. The standard treatments for diabetic patients are glucose and blood pressure control, lipid lowering, and renin-angiotensin system blockade; however, these therapeutic approaches can provide only partial renoprotection if started late in the course of the disease. One major limitation in developing efficient therapies for DN is the complex pathobiology of the diabetic kidney, which undergoes a set of profound structural, metabolic and functional changes. Despite these difficulties, experimental models of diabetes have revealed promising therapeutic targets by identifying pathways that modulate key functions of podocytes and glomerular endothelial cells. In this review we will describe recent advances in the field, analyze key molecular pathways that contribute to the pathogenesis of the disease, and discuss how they could be modulated to prevent or reverse DN.
Keywords: angiotensin 1–7; diabetic nephropathy; hypoxia inducible factor; notch signaling; renin-angiotensin system; sirtuins; sodium-glucose cotransporter 2; thyroid hormone signaling.
Copyright © 2020 Zoja, Xinaris and Macconi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

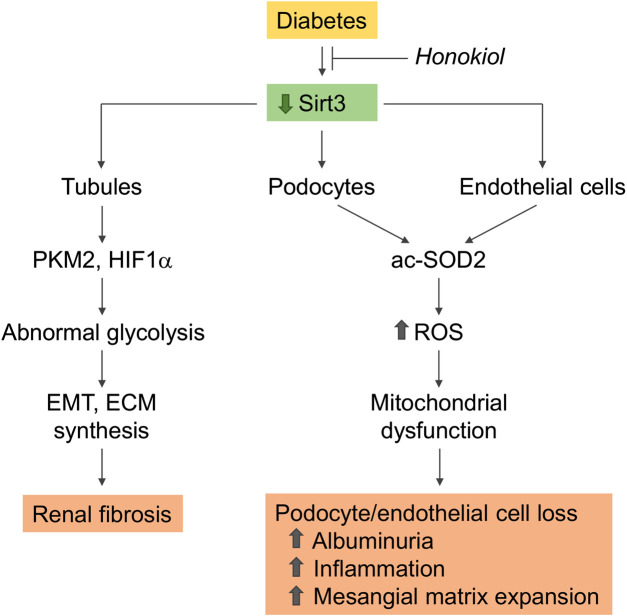
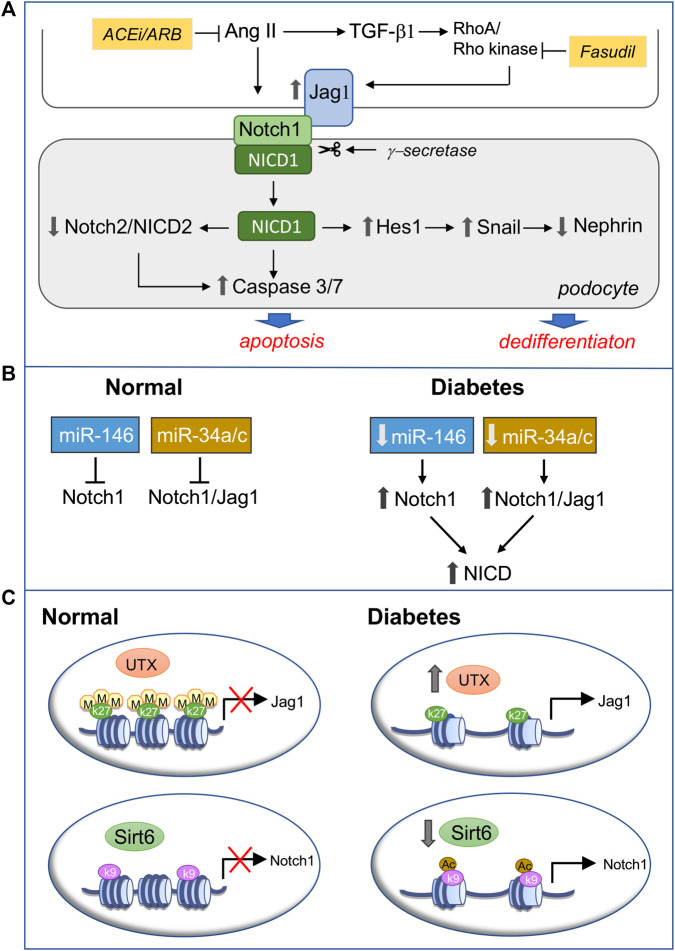
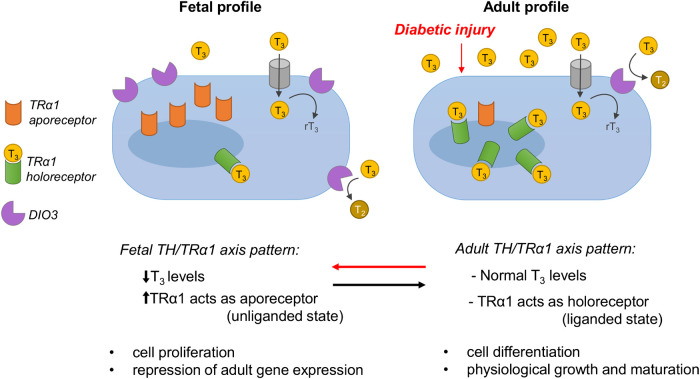
References
Publication types
LinkOut - more resources
Full Text Sources