

VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses
- PMID: 33522966
- PMCID: PMC7852108
- DOI: 10.1186/s40168-020-00990-y
VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses
Abstract
Background: Viruses are a significant player in many biosphere and human ecosystems, but most signals remain "hidden" in metagenomic/metatranscriptomic sequence datasets due to the lack of universal gene markers, database representatives, and insufficiently advanced identification tools.
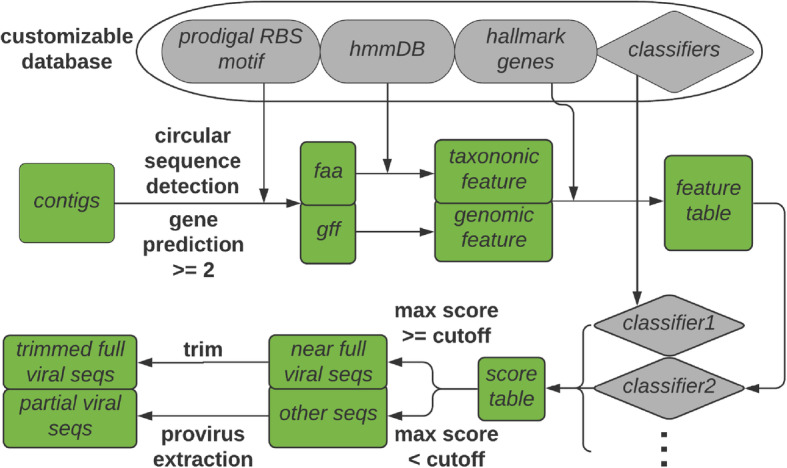
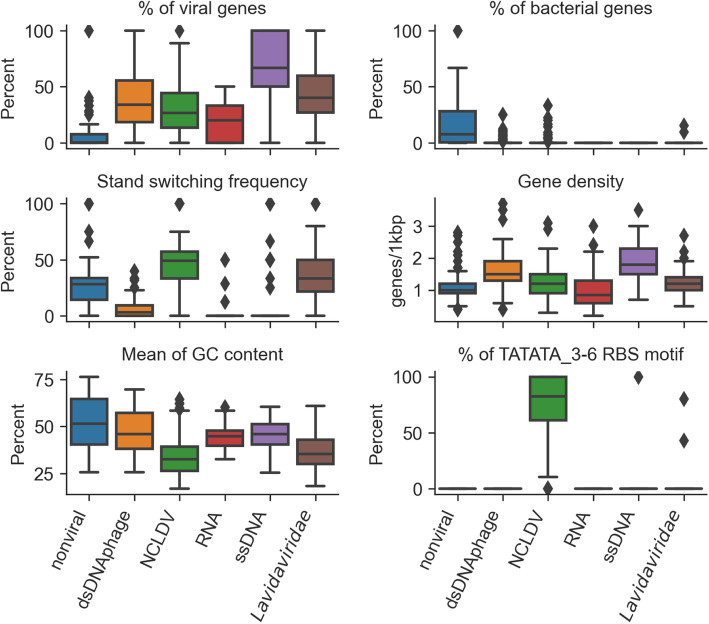
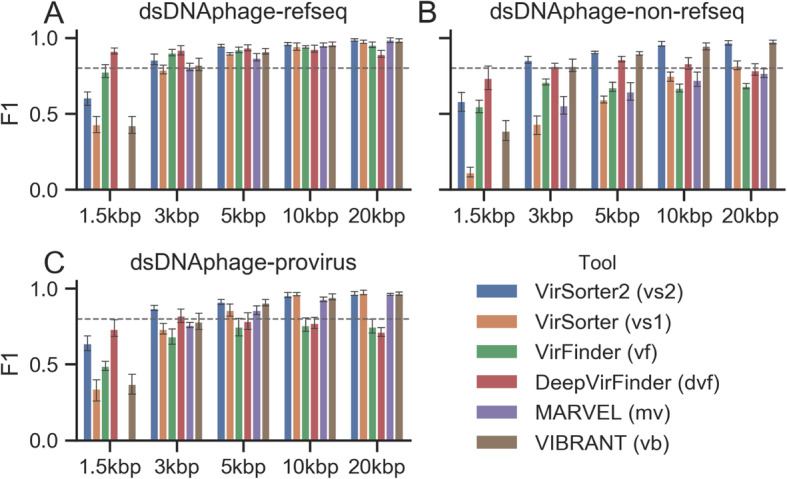
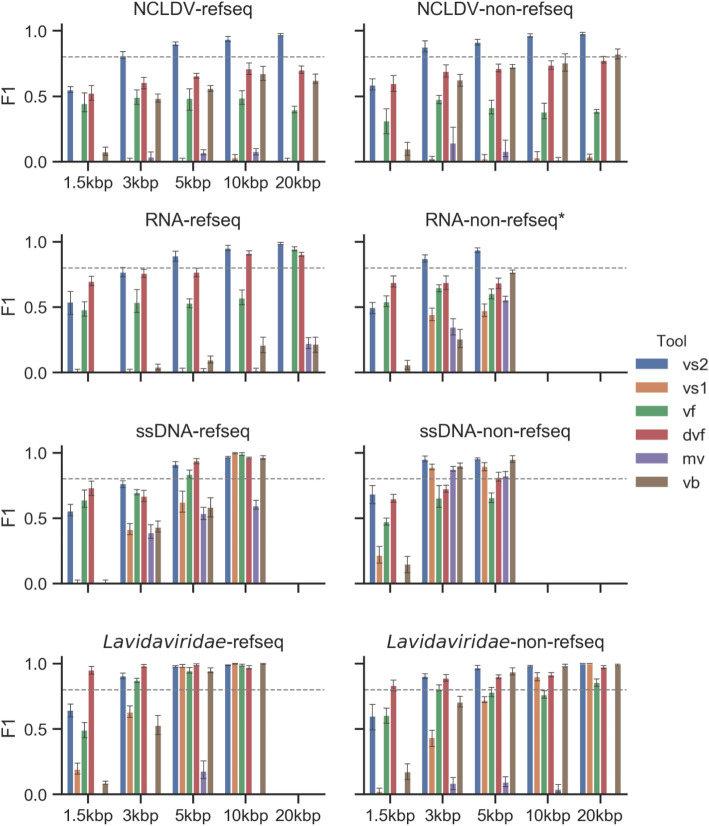
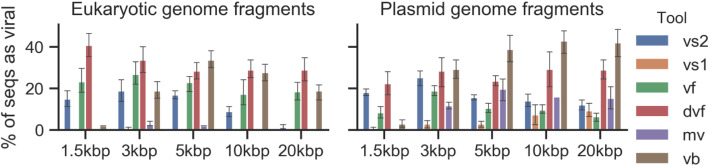
Results: Here, we introduce VirSorter2, a DNA and RNA virus identification tool that leverages genome-informed database advances across a collection of customized automatic classifiers to improve the accuracy and range of virus sequence detection. When benchmarked against genomes from both isolated and uncultivated viruses, VirSorter2 uniquely performed consistently with high accuracy (F1-score > 0.8) across viral diversity, while all other tools under-detected viruses outside of the group most represented in reference databases (i.e., those in the order Caudovirales). Among the tools evaluated, VirSorter2 was also uniquely able to minimize errors associated with atypical cellular sequences including eukaryotic genomes and plasmids. Finally, as the virosphere exploration unravels novel viral sequences, VirSorter2's modular design makes it inherently able to expand to new types of viruses via the design of new classifiers to maintain maximal sensitivity and specificity.
Conclusion: With multi-classifier and modular design, VirSorter2 demonstrates higher overall accuracy across major viral groups and will advance our knowledge of virus evolution, diversity, and virus-microbe interaction in various ecosystems. Source code of VirSorter2 is freely available ( https://bitbucket.org/MAVERICLab/virsorter2 ), and VirSorter2 is also available both on bioconda and as an iVirus app on CyVerse ( https://de.cyverse.org/de ). Video abstract.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
