

NEDD9 Is a Novel and Modifiable Mediator of Platelet-Endothelial Adhesion in the Pulmonary Circulation
- PMID: 33523764
- PMCID: PMC8483217
- DOI: 10.1164/rccm.202003-0719OC
NEDD9 Is a Novel and Modifiable Mediator of Platelet-Endothelial Adhesion in the Pulmonary Circulation
Abstract
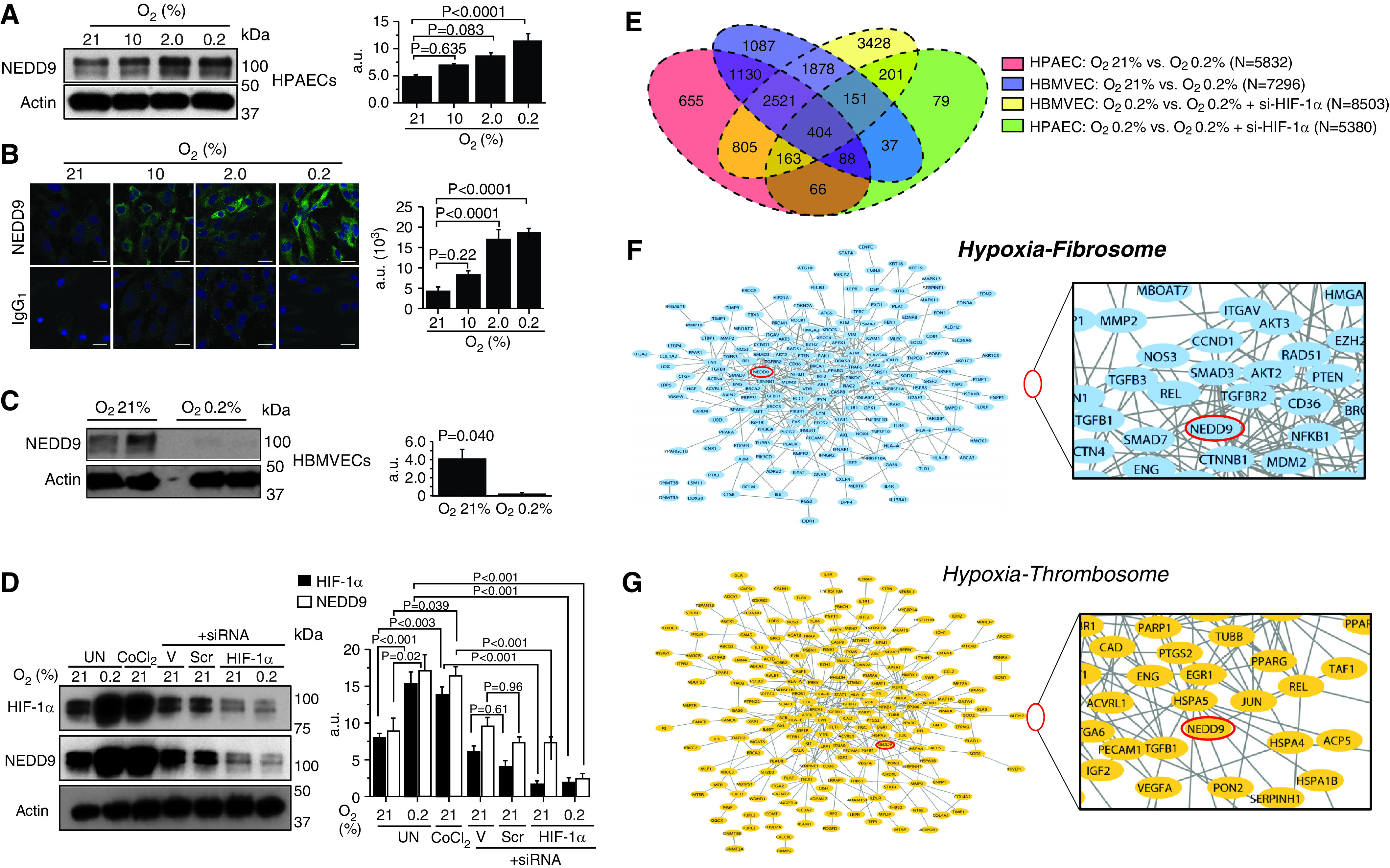
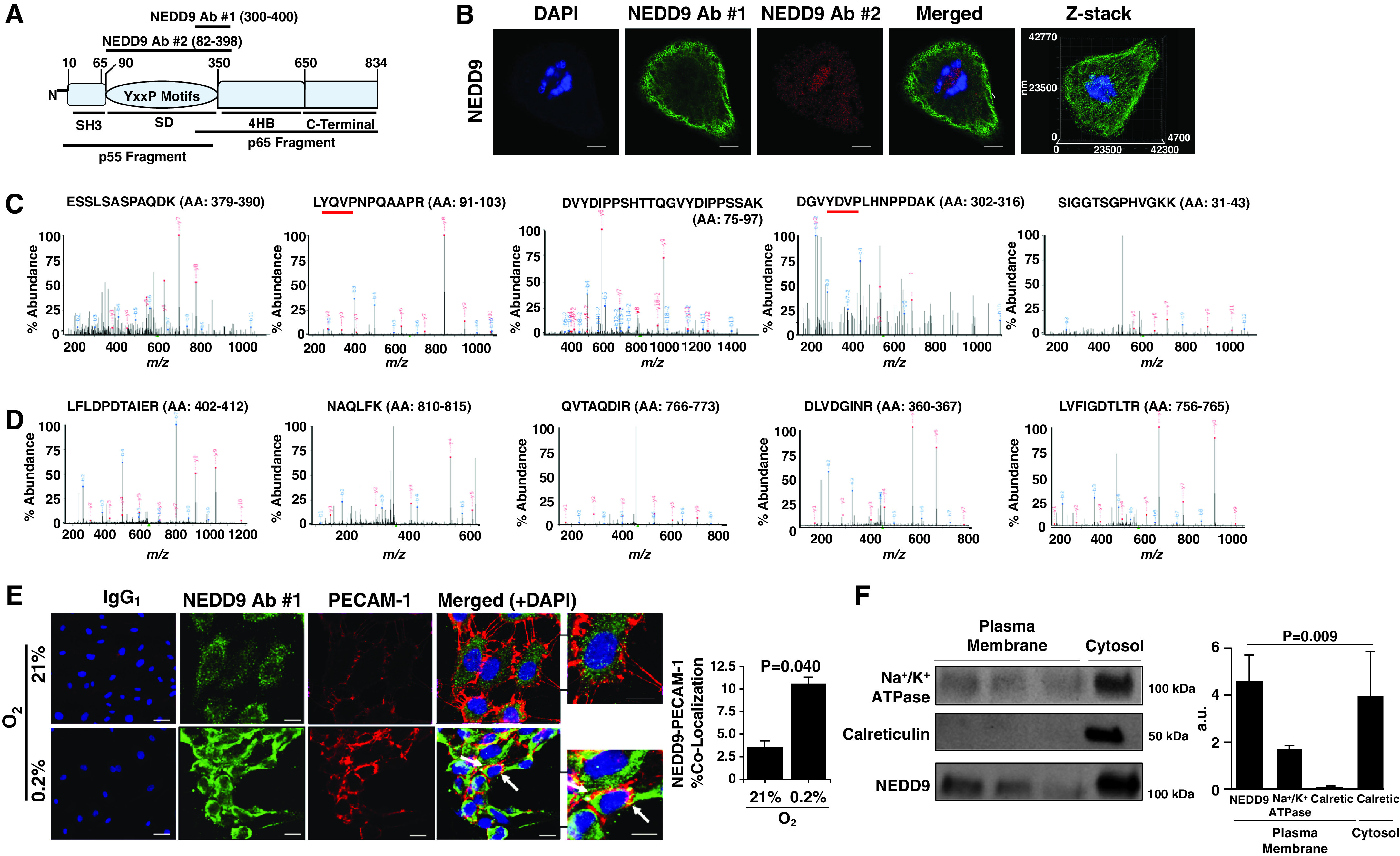
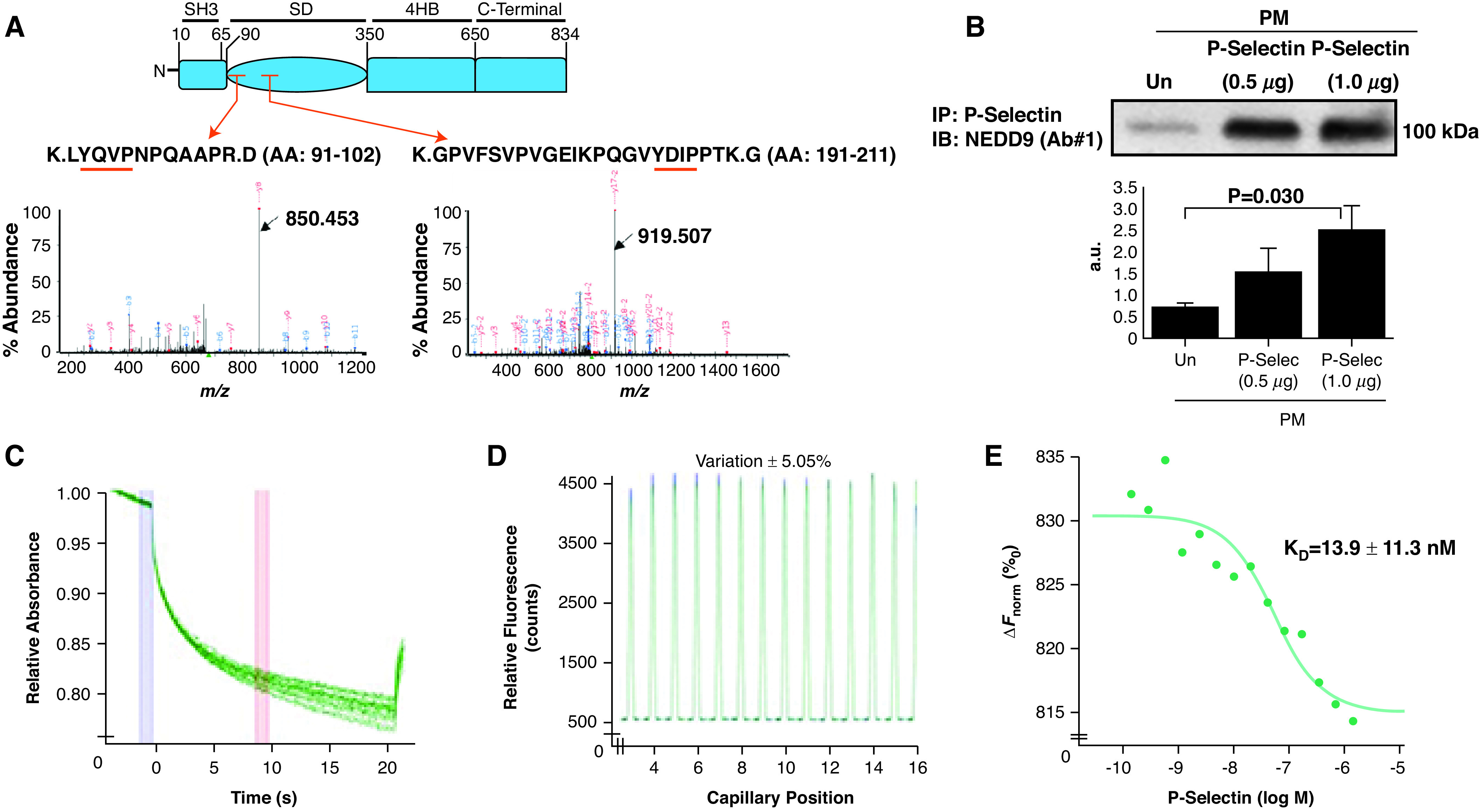
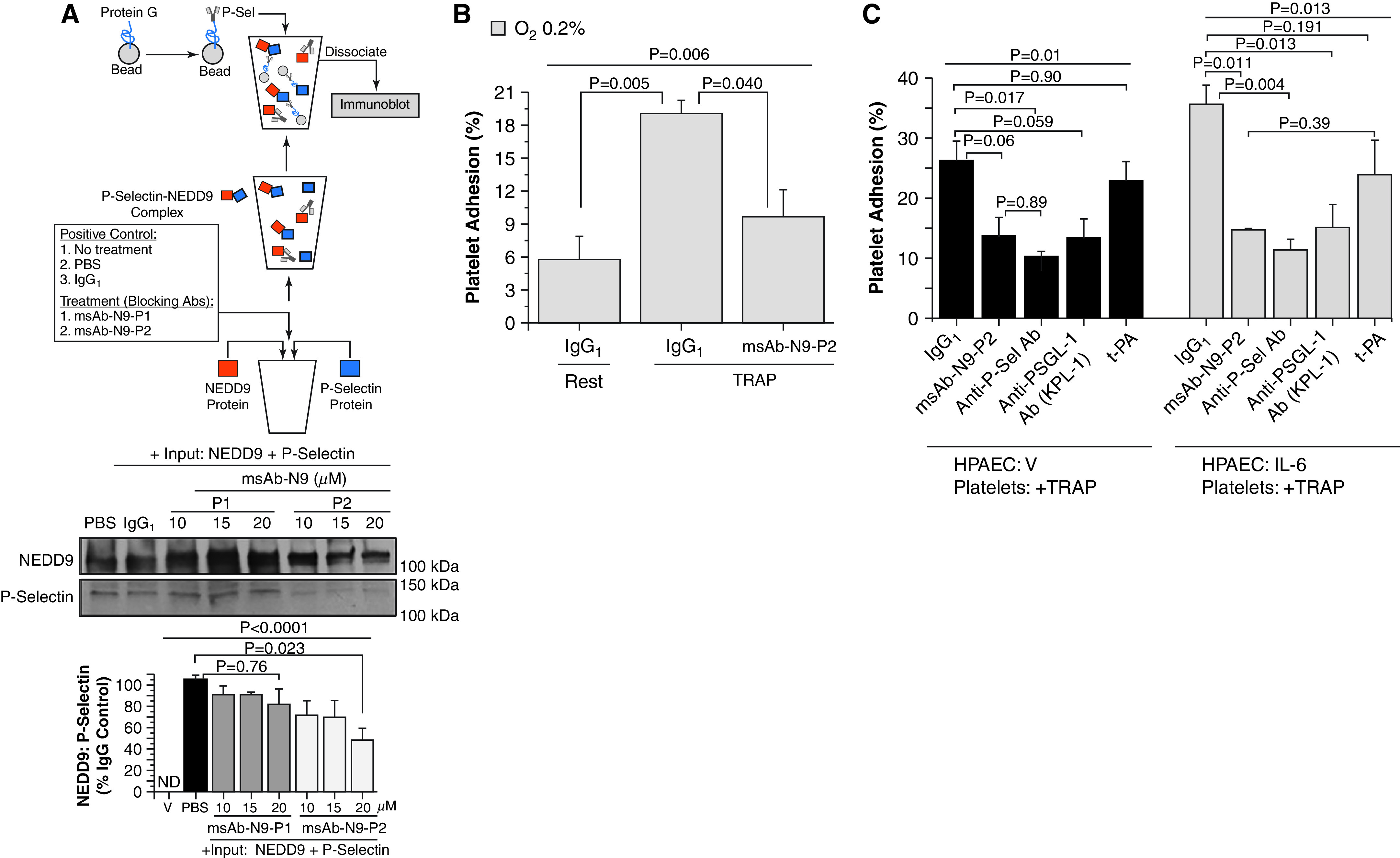
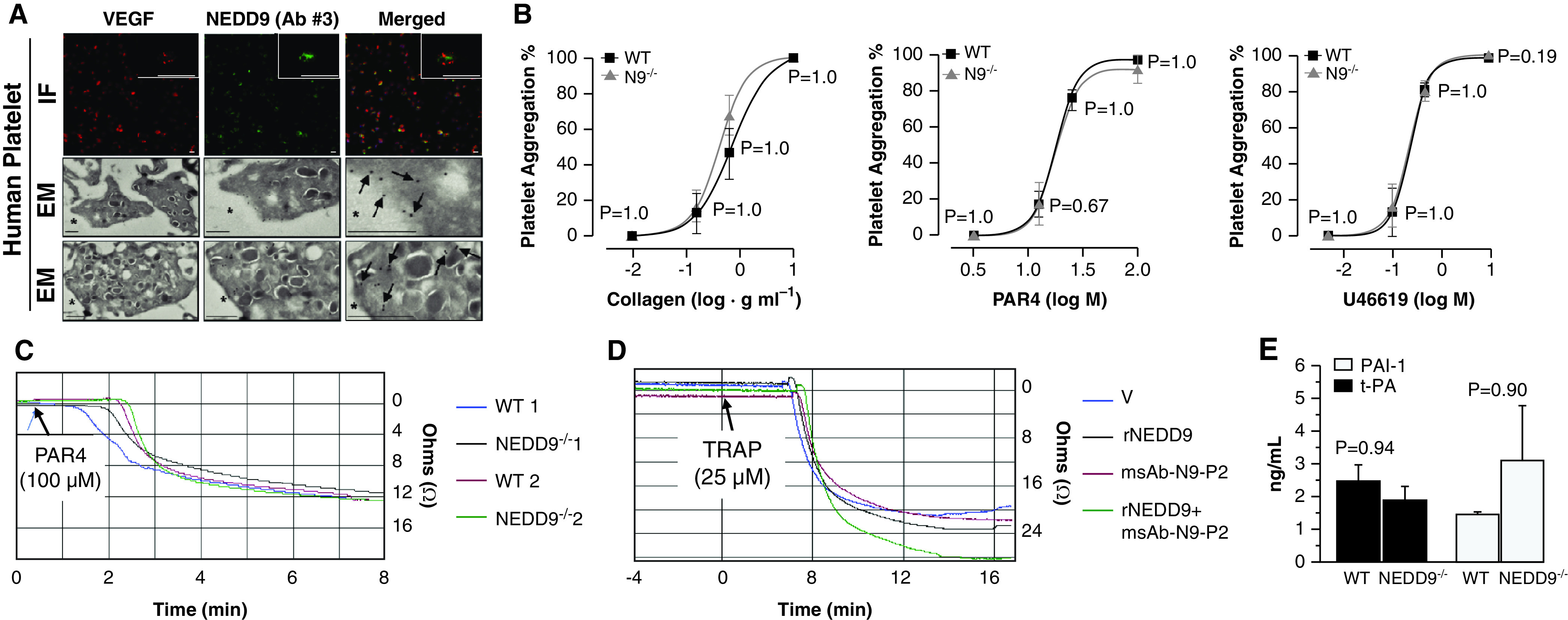
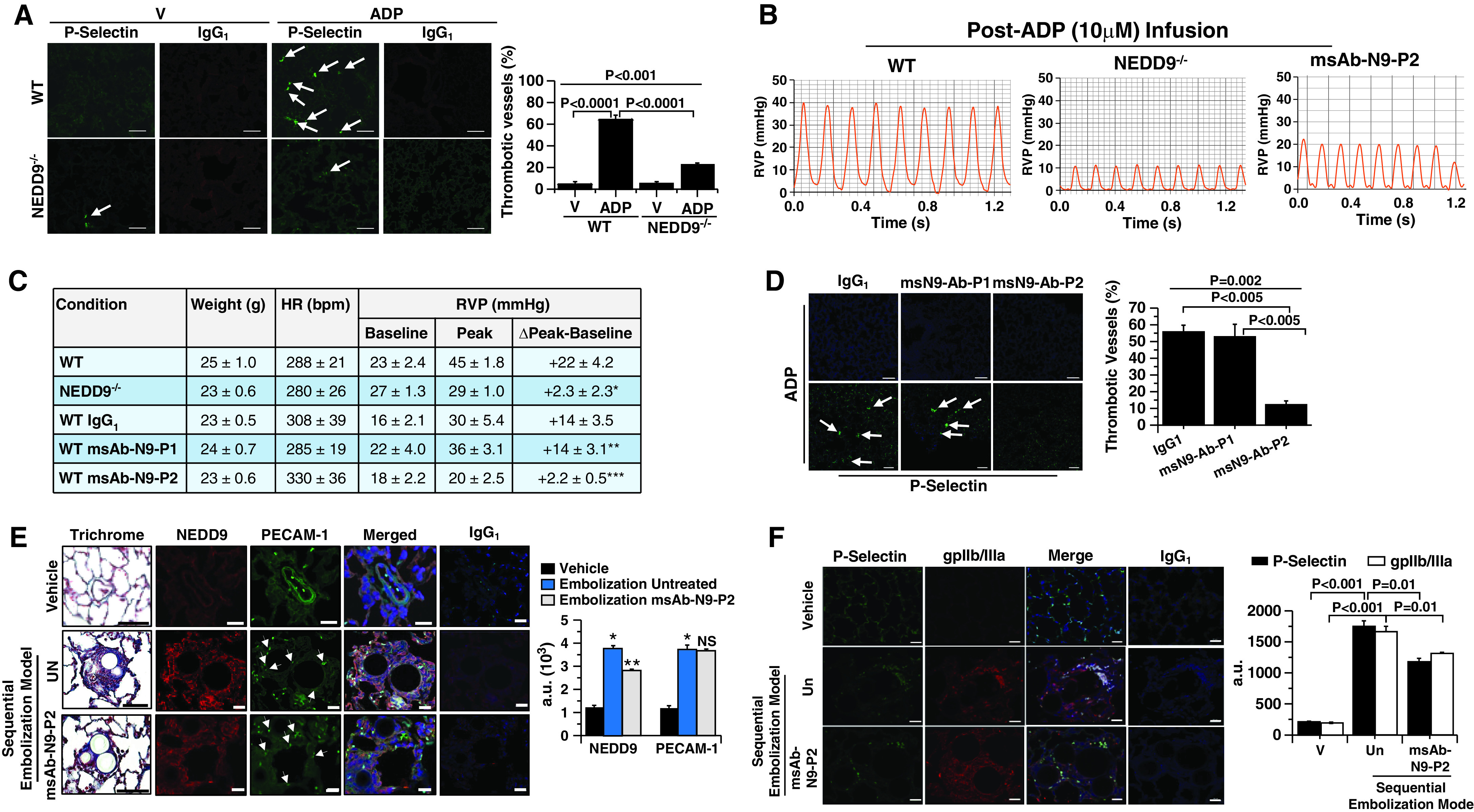
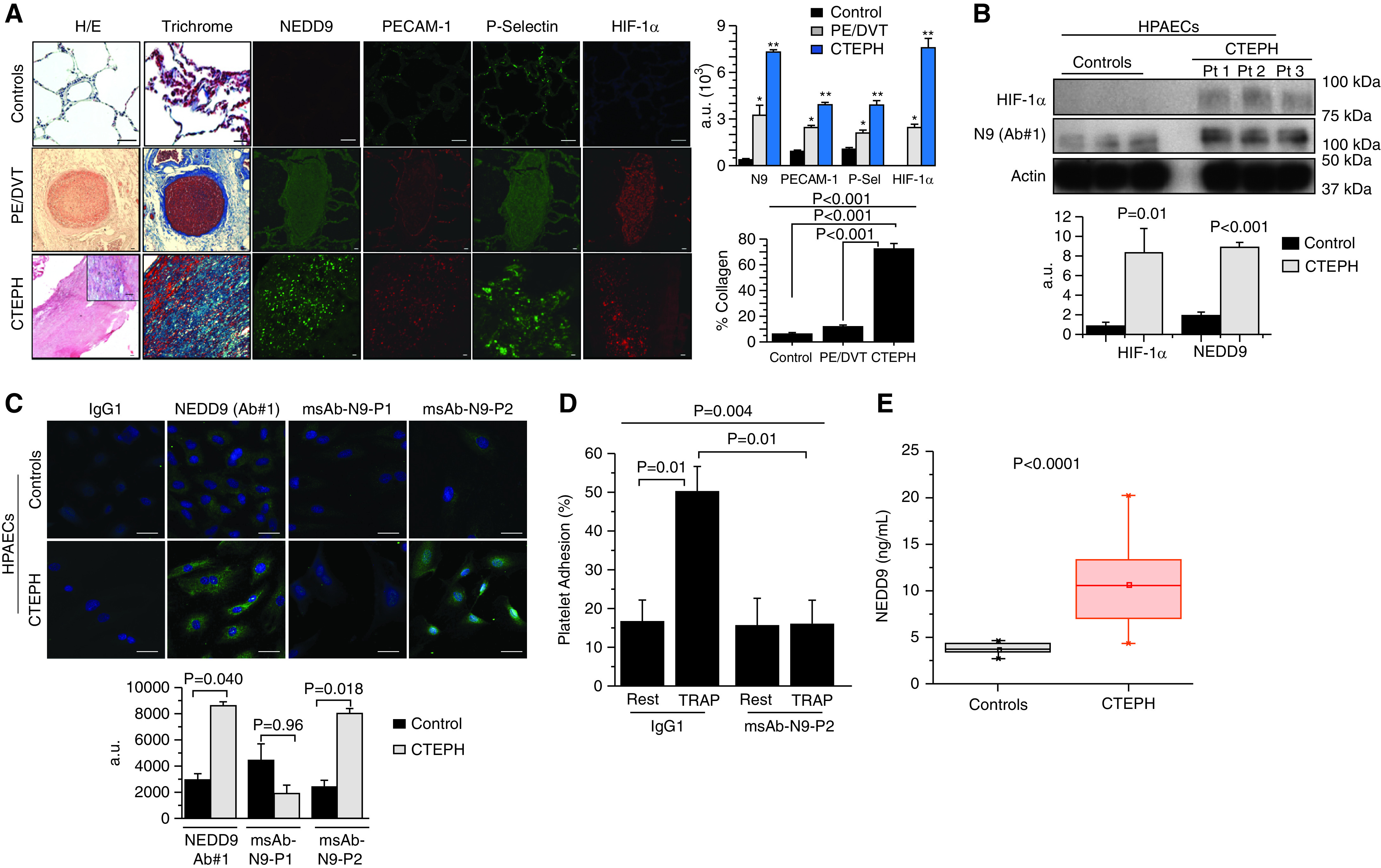
Rationale: Data on the molecular mechanisms that regulate platelet-pulmonary endothelial adhesion under conditions of hypoxia are lacking, but may have important therapeutic implications. Objectives: To identify a hypoxia-sensitive, modifiable mediator of platelet-pulmonary artery endothelial cell adhesion and thrombotic remodeling. Methods: Network medicine was used to profile protein-protein interactions in hypoxia-treated human pulmonary artery endothelial cells. Data from liquid chromatography-mass spectrometry and microscale thermophoresis informed the development of a novel antibody (Ab) to inhibit platelet-endothelial adhesion, which was tested in cells from patients with chronic thromboembolic pulmonary hypertension (CTEPH) and three animal models in vivo. Measurements and Main Results: The protein NEDD9 was identified in the hypoxia thrombosome network in silico. Compared with normoxia, hypoxia (0.2% O2) for 24 hours increased HIF-1α (hypoxia-inducible factor-1α)-dependent NEDD9 upregulation in vitro. Increased NEDD9 was localized to the plasma-membrane surface of cells from control donors and patients with CTEPH. In endarterectomy specimens, NEDD9 colocalized with the platelet surface adhesion molecule P-selectin. Our custom-made anti-NEDD9 Ab targeted the NEDD9-P-selectin interaction and inhibited the adhesion of activated platelets to pulmonary artery endothelial cells from control donors in vitro and from patients with CTEPH ex vivo. Compared with control mice, platelet-pulmonary endothelial aggregates and pulmonary hypertension induced by ADP were decreased in NEDD9-/- mice or wild-type mice treated with the anti-NEDD9 Ab, which also decreased chronic pulmonary thromboembolic remodeling in vivo. Conclusions: The NEDD9-P-selectin protein-protein interaction is a modifiable target with which to inhibit platelet-pulmonary endothelial adhesion and thromboembolic vascular remodeling, with potential therapeutic implications for patients with disorders of increased hypoxia signaling pathways, including CTEPH.
Keywords: hypoxia; platelets; thrombosis.
Figures
Comment in
-
NEDD9, a Hypoxia-upregulated Mediator for Pathogenic Platelet-Endothelial Cell Interaction in Pulmonary Hypertension.Am J Respir Crit Care Med. 2021 Jun 15;203(12):1455-1458. doi: 10.1164/rccm.202101-0007ED. Am J Respir Crit Care Med. 2021. PMID: 33770456 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- U01 HG007690/HG/NHGRI NIH HHS/United States
- T32 HL007633/HL/NHLBI NIH HHS/United States
- KL2 TR002542/TR/NCATS NIH HHS/United States
- R21 HL145420/HL/NHLBI NIH HHS/United States
- U54 HL119145/HL/NHLBI NIH HHS/United States
- T32 GM136444/GM/NIGMS NIH HHS/United States
- R03 HL158640/HL/NHLBI NIH HHS/United States
- R01 HL139613/HL/NHLBI NIH HHS/United States
- R56 HL131787/HL/NHLBI NIH HHS/United States
- R01 HL061795/HL/NHLBI NIH HHS/United States
- K08 HL121185/HL/NHLBI NIH HHS/United States
- P50 GM107618/GM/NIGMS NIH HHS/United States
- R01 HL153502/HL/NHLBI NIH HHS/United States
- T32 CA009216/CA/NCI NIH HHS/United States
- R37 HL061795/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
