

Synthetic lethality across normal tissues is strongly associated with cancer risk, onset, and tumor suppressor specificity
- PMID: 33523837
- PMCID: PMC7775773
- DOI: 10.1126/sciadv.abc2100
Synthetic lethality across normal tissues is strongly associated with cancer risk, onset, and tumor suppressor specificity
Abstract
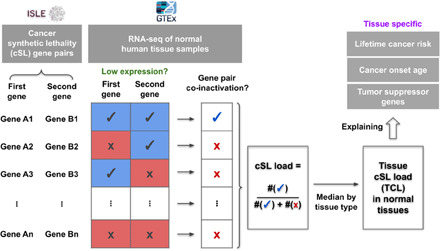
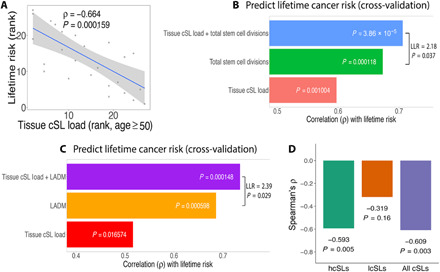
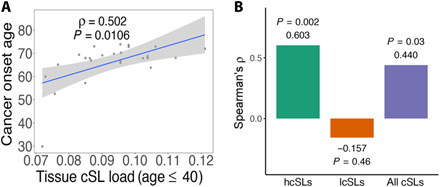
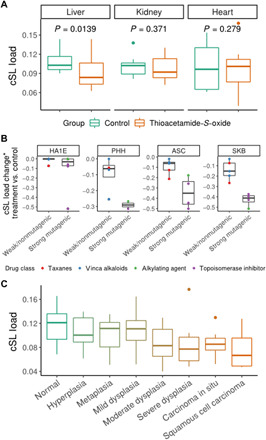
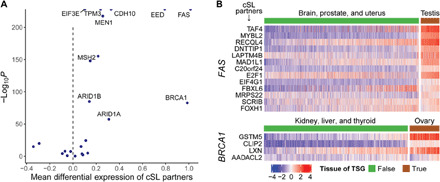
Various characteristics of cancers exhibit tissue specificity, including lifetime cancer risk, onset age, and cancer driver genes. Previously, the large variation in cancer risk across human tissues was found to strongly correlate with the number of stem cell divisions and abnormal DNA methylation levels. Here, we study the role of synthetic lethality in cancer risk. Analyzing normal tissue transcriptomics data in the Genotype-Tissue Expression project, we quantify the extent of co-inactivation of cancer synthetic lethal (cSL) gene pairs and find that normal tissues with more down-regulated cSL gene pairs have lower and delayed cancer risk. Consistently, more cSL gene pairs become up-regulated in cells treated by carcinogens and throughout premalignant stages in vivo. We also show that the tissue specificity of numerous tumor suppressor genes is associated with the expression of their cSL partner genes across normal tissues. Overall, our findings support the possible role of synthetic lethality in tumorigenesis.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Surveillance, Epidemiology, and End Results (SEER) Program, “SEER*Stat Database Incidence - SEER 9 Regs Research Data, Nov 2017 Sub (1973–2015) - Linked To County Attributes - Total U.S., 1969–2016 Counties,” National Cancer Institute, DCCPS, Surveillance Research Program, released April 2018, based on the November 2017 submission (2018); www.seer.cancer.gov.
-
- Feinberg A. P., Ohlsson R., Henikoff S., The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33 (2006). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
