

BBB pathophysiology-independent delivery of siRNA in traumatic brain injury
- PMID: 33523853
- PMCID: PMC7775748
- DOI: 10.1126/sciadv.abd6889
BBB pathophysiology-independent delivery of siRNA in traumatic brain injury
Abstract
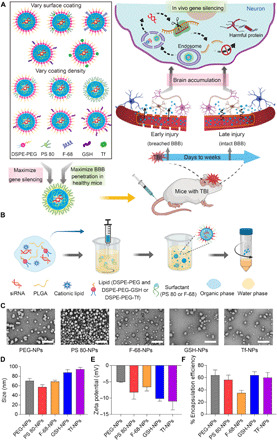
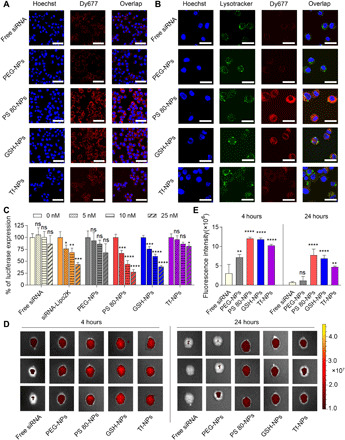
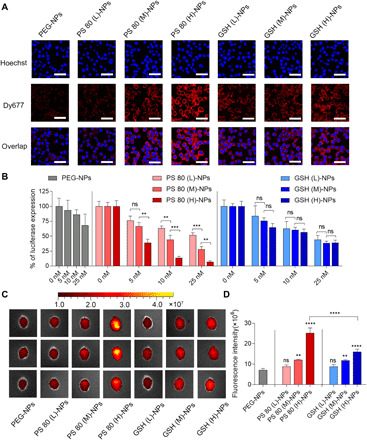
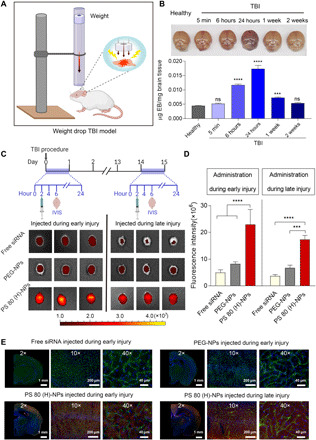
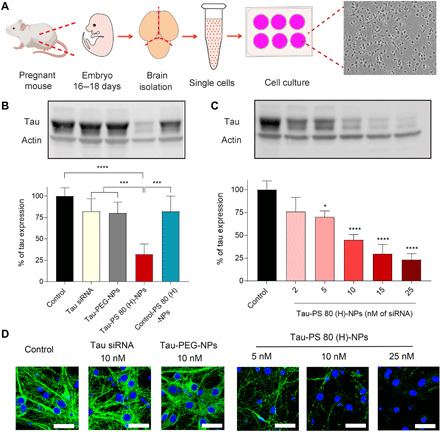
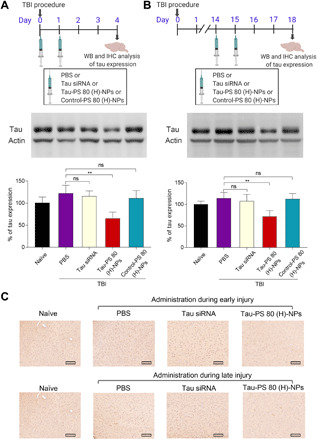
Small interfering RNA (siRNA)-based therapeutics can mitigate the long-term sequelae of traumatic brain injury (TBI) but suffer from poor permeability across the blood-brain barrier (BBB). One approach to overcoming this challenge involves treatment administration while BBB is transiently breached after injury. However, it offers a limited window for therapeutic intervention and is applicable to only a subset of injuries with substantially breached BBB. We report a nanoparticle platform for BBB pathophysiology-independent delivery of siRNA in TBI. We achieved this by combined modulation of surface chemistry and coating density on nanoparticles, which maximized their active transport across BBB. Engineered nanoparticles injected within or outside the window of breached BBB in TBI mice showed threefold higher brain accumulation compared to nonengineered PEGylated nanoparticles and 50% gene silencing. Together, our data suggest that this nanoparticle platform is a promising next-generation drug delivery approach for the treatment of TBI.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Roozenbeek B., Maas A. I. R., Menon D. K., Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 9, 231–236 (2013). - PubMed
-
- Meabon J. S., Huber B. R., Cross D. J., Richards T. L., Minoshima S., Pagulayan K. F., Li G., Meeker K. D., Kraemer B. C., Petrie E. C., Raskind M. A., Peskind E. R., Cook D. G., Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci. Transl. Med. 8, 321ra6 (2016). - PubMed
-
- Goldstein L. E., Fisher A. M., Tagge C. A., Zhang X.-L., Velisek L., Sullivan J. A., Upreti C., Kracht J. M., Ericsson M., Wojnarowicz M. W., Goletiani C. J., Maglakelidze G. M., Casey N., Moncaster J. A., Minaeva O., Moir R. D., Nowinski C. J., Stern R. A., Cantu R. C., Geiling J., Blusztajn J. K., Wolozin B. L., Ikezu T., Stein T. D., Budson A. E., Kowall N. W., Chargin D., Sharon A., Saman S., Hall G. F., Moss W. C., Cleveland R. O., Tanzi R. E., Stanton P. K., McKee A. C., Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, 134ra160 (2012). - PMC - PubMed
-
- McKee A. C., Stein D., Nowinski C. J., Stern R. A., Daneshvar D. H., Alvarez V. E., Lee H.-S., Hall G., Wojtowicz S. M., Baugh C. M., Riley D. O., Kubilus C. A., Cormier K. A., Jacobs M. A., Martin B. R., Abraham C. R., Ikezu T., Reichard R. R., Wolozin B. L., Budson A. E., Goldstein L. E., Kowall N. W., Cantu R. C., The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64 (2013). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
