

Recessive NOS1AP variants impair actin remodeling and cause glomerulopathy in humans and mice
- PMID: 33523862
- PMCID: PMC10763988
- DOI: 10.1126/sciadv.abe1386
Recessive NOS1AP variants impair actin remodeling and cause glomerulopathy in humans and mice
Abstract
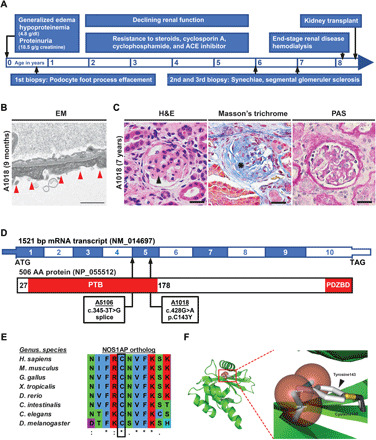
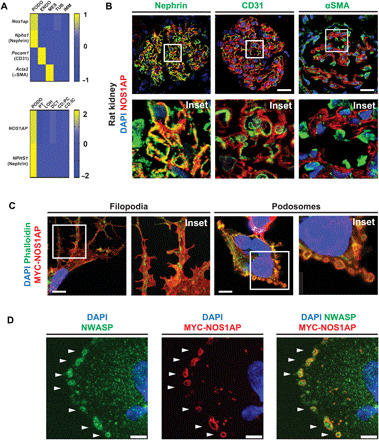
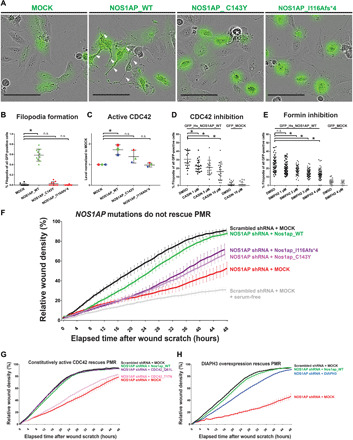
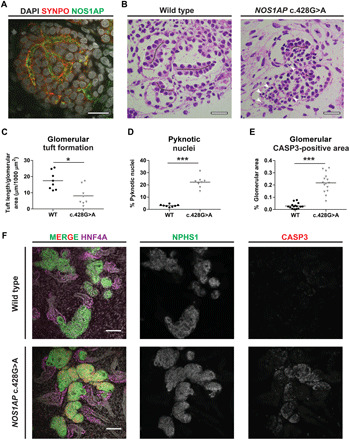
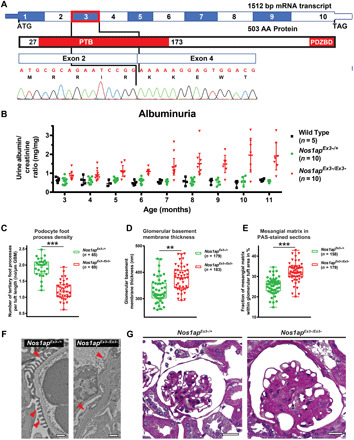
Nephrotic syndrome (NS) is a leading cause of chronic kidney disease. We found recessive NOS1AP variants in two families with early-onset NS by exome sequencing. Overexpression of wild-type (WT) NOS1AP, but not cDNA constructs bearing patient variants, increased active CDC42 and promoted filopodia and podosome formation. Pharmacologic inhibition of CDC42 or its effectors, formin proteins, reduced NOS1AP-induced filopodia formation. NOS1AP knockdown reduced podocyte migration rate (PMR), which was rescued by overexpression of WT Nos1ap but not by constructs bearing patient variants. PMR in NOS1AP knockdown podocytes was also rescued by constitutively active CDC42Q61L or the formin DIAPH3 Modeling a NOS1AP patient variant in knock-in human kidney organoids revealed malformed glomeruli with increased apoptosis. Nos1apEx3-/Ex3- mice recapitulated the human phenotype, exhibiting proteinuria, foot process effacement, and glomerulosclerosis. These findings demonstrate that recessive NOS1AP variants impair CDC42/DIAPH-dependent actin remodeling, cause aberrant organoid glomerulogenesis, and lead to a glomerulopathy in humans and mice.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- W. Harmon, R. Fine, S. Alexander, B. Warady, M. Benfield, S. Goldstein, R. McDonald, K. Martz, D. Stablein, NAPRTCS 2008 Annual Report (2008).
-
- Wiggins R.-C., The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 71, 1205–1214 (2007). - PubMed
-
- Connaughton D. M., Kennedy C., Shril S., Mann N., Murray S. L., Williams P. A., Conlon E., Nakayama M., van der Ven A. T., Ityel H., Kause F., Kolvenbach C. M., Dai R., Vivante A., Braun D. A., Schneider R., Kitzler T. M., Moloney B., Moran C. P., Smyth J. S., Kennedy A., Benson K., Stapleton C., Denton M., Magee C., O’Seaghdha C. M., Plant W. D., Griffin M. D., Awan A., Sweeney C., Mane S. M., Lifton R. P., Griffin B., Leavey S., Casserly L., de Freitas D. G., Holian J., Dorman A., Doyle B., Lavin P. J., Little M. A., Conlon P. J., Hildebrandt F., Monogenic causes of chronic kidney disease in adults. Kidney Int. 95, 914–928 (2019). - PMC - PubMed
-
- Sadowski C. E., Lovric S., Ashraf S., Pabst W. L., Gee H. Y., Kohl S., Engelmann S., Vega-Warner V., Fang H., Halbritter J., Somers M. J., Tan W., Shril S., Fessi I., Lifton R. P., Bockenhauer D., El-Desoky S., Kari J. A., Zenker M., Kemper M. J., Mueller D., Fathy H. M., Soliman N. A.; SRNS Study Group and Friedhelm Hildebrandt , A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 26, 1279–1289 (2015). - PMC - PubMed
-
- Warejko J. K., Tan W., Daga A., Schapiro D., Lawson J. A., Shril S., Lovric S., Ashraf S., Rao J., Hermle T., Jobst-Schwan T., Widmeier E., Majmundar A. J., Schneider R., Gee H. Y., Schmidt J. M., Vivante A., van der Ven A. T., Ityel H., Chen J., Sadowski C. E., Kohl S., Pabst W. L., Nakayama M., Somers M. J. G., Rodig N. M., Daouk G., Baum M., Stein D. R., Ferguson M. A., Traum A. Z., Soliman N. A., Kari J. A., Desoky S. E., Fathy H., Zenker M., Bakkaloglu S. A., Müller D., Noyan A., Ozaltin F., Cadnapaphornchai M. A., Hashmi S., Hopcian J., Kopp J. B., Benador N., Bockenhauer D., Bogdanovic R., Stajić N., Chernin G., Ettenger R., Fehrenbach H., Kemper M., Munarriz R. L., Podracka L., Büscher R., Serdaroglu E., Tasic V., Mane S., Lifton R. P., Braun D. A., Hildebrandt F., Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 13, 53–62 (2018). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
