

Multiple screening approaches reveal HDAC6 as a novel regulator of glycolytic metabolism in triple-negative breast cancer
- PMID: 33523897
- PMCID: PMC7810372
- DOI: 10.1126/sciadv.abc4897
Multiple screening approaches reveal HDAC6 as a novel regulator of glycolytic metabolism in triple-negative breast cancer
Abstract
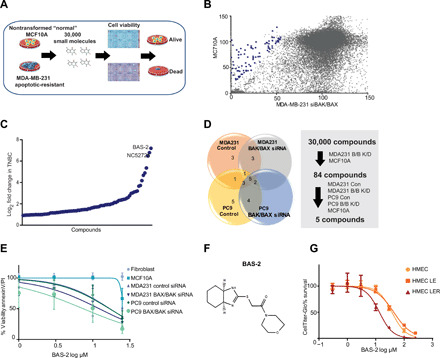
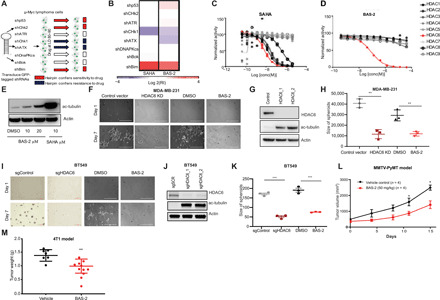
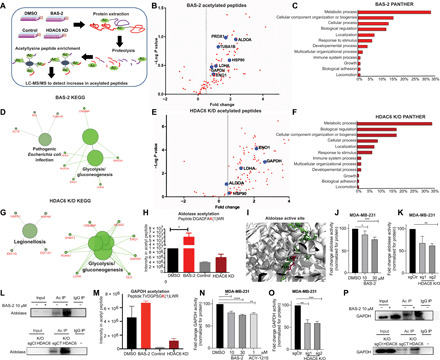
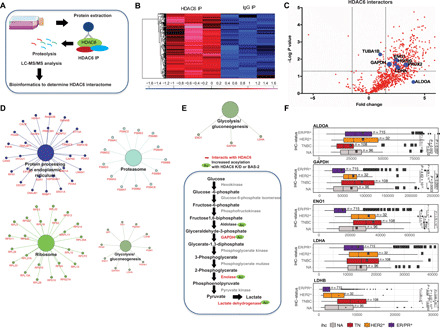
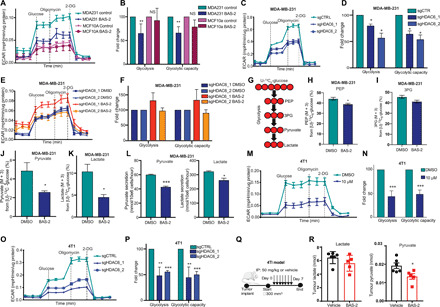
Triple-negative breast cancer (TNBC) is a subtype of breast cancer without a targeted form of therapy. Unfortunately, up to 70% of patients with TNBC develop resistance to treatment. A known contributor to chemoresistance is dysfunctional mitochondrial apoptosis signaling. We set up a phenotypic small-molecule screen to reveal vulnerabilities in TNBC cells that were independent of mitochondrial apoptosis. Using a functional genetic approach, we identified that a "hit" compound, BAS-2, had a potentially similar mechanism of action to histone deacetylase inhibitors (HDAC). An in vitro HDAC inhibitor assay confirmed that the compound selectively inhibited HDAC6. Using state-of-the-art acetylome mass spectrometry, we identified glycolytic substrates of HDAC6 in TNBC cells. We confirmed that inhibition or knockout of HDAC6 reduced glycolytic metabolism both in vitro and in vivo. Through a series of unbiased screening approaches, we have identified a previously unidentified role for HDAC6 in regulating glycolytic metabolism.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution License 4.0 (CC BY).
Figures
References
-
- Perou C. M., Sørlie T., Eisen M. B., van de Rijn M., Jeffrey S. S., Rees C. A., Pollack J. R., Ross D. T., Johnsen H., Akslen L. A., Fluge Ø., Pergamenschikov A., Williams C., Zhu S. X., Lønning P. E., Børresen-Dale A.-L., Brown P. O., Botstein D., Molecular portraits of human breast tumours. Nature 406, 747–752 (2000). - PubMed
-
- Carey L., Winer E., Viale G., Cameron D., Gianni L., Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 7, 683–692 (2010). - PubMed
-
- Liedtke C., Mazouni C., Hess K. R., André F., Tordai A., Mejia J. A., Symmans W. F., Gonzalez-Angulo A. M., Hennessy B., Green M., Cristofanilli M., Hortobagyi G. N., Pusztai L., Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 26, 1275–1281 (2008). - PubMed
-
- Holohan C., Van Schaeybroeck S., Longley D. B., Johnston P. G., Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
