

Linkage-specific deubiquitylation by OTUD5 defines an embryonic pathway intolerant to genomic variation
- PMID: 33523931
- PMCID: PMC7817106
- DOI: 10.1126/sciadv.abe2116
Linkage-specific deubiquitylation by OTUD5 defines an embryonic pathway intolerant to genomic variation
Abstract
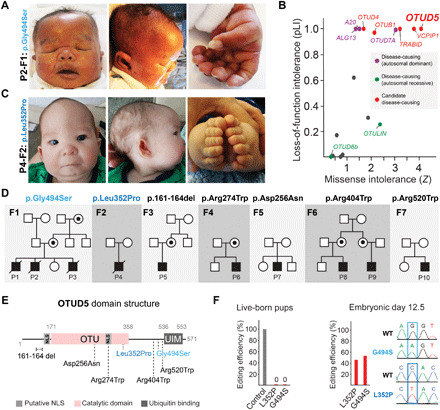
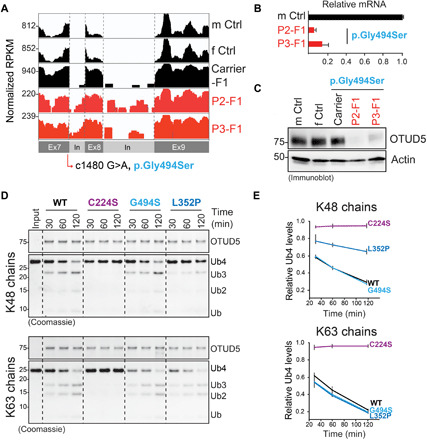
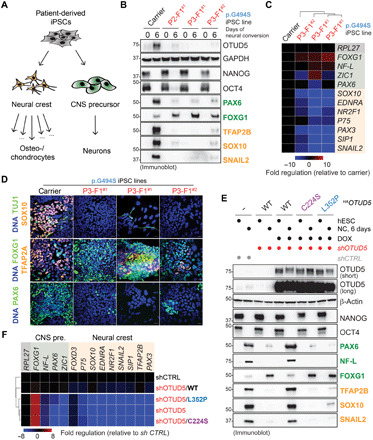
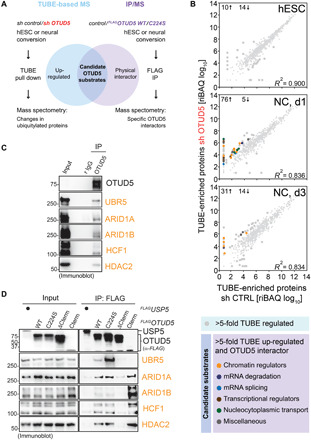
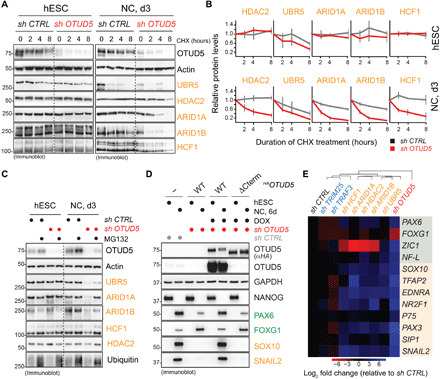
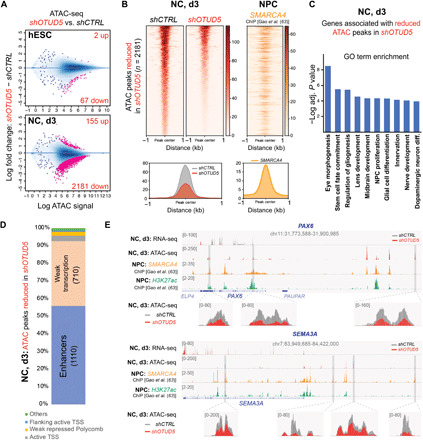
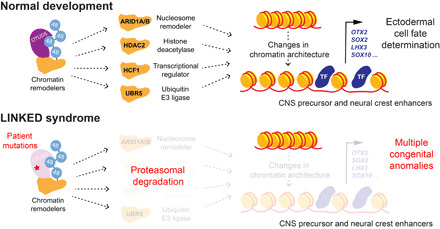
Reversible modification of proteins with linkage-specific ubiquitin chains is critical for intracellular signaling. Information on physiological roles and underlying mechanisms of particular ubiquitin linkages during human development are limited. Here, relying on genomic constraint scores, we identify 10 patients with multiple congenital anomalies caused by hemizygous variants in OTUD5, encoding a K48/K63 linkage-specific deubiquitylase. By studying these mutations, we find that OTUD5 controls neuroectodermal differentiation through cleaving K48-linked ubiquitin chains to counteract degradation of select chromatin regulators (e.g., ARID1A/B, histone deacetylase 2, and HCF1), mutations of which underlie diseases that exhibit phenotypic overlap with OTUD5 patients. Loss of OTUD5 during differentiation leads to less accessible chromatin at neuroectodermal enhancers and aberrant gene expression. Our study describes a previously unidentified disorder we name LINKED (LINKage-specific deubiquitylation deficiency-induced Embryonic Defects) syndrome and reveals linkage-specific ubiquitin cleavage from chromatin remodelers as an essential signaling mode that coordinates chromatin remodeling during embryogenesis.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Claussnitzer M., Cho J. H., Collins R., Cox N. J., Dermitzakis E. T., Hurles M. E., Kathiresan S., Kenny E. E., Lindgren C. M., MacArthur D. G., North K. N., Plon S. E., Rehm H. L., Risch N., Rotimi C. N., Shendure J., Soranzo N., McCarthy M. I., A brief history of human disease genetics. Nature 577, 179–189 (2020). - PMC - PubMed
-
- Wright C. F., FitzPatrick D. R., Firth H. V., Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet. 19, 253–268 (2018). - PubMed
-
- Komander D., Rape M., The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (2012). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
