

Cystic Fibrosis
- PMID: 33526571
- PMCID: PMC8972143
- DOI: 10.1542/pir.2019-0212
Cystic Fibrosis
Abstract
Cystic fibrosis (CF) is one of the most commonly diagnosed genetic disorders. Clinical characteristics include progressive obstructive lung disease, sinusitis, exocrine pancreatic insufficiency leading to malabsorption and malnutrition, liver and pancreatic dysfunction, and male infertility. Although CF is a life-shortening disease, survival has continued to improve to a median age of 46.2 years due to earlier diagnosis through routine newborn screening, promulgation of evidence-based guidelines to optimize nutritional and pulmonary health, and the development of CF-specific interdisciplinary care centers. Future improvements in health and quality of life for individuals with CF are likely with the recent development of mutation-specific modulator therapies. In this review, we will cover the current understanding of the disease manifestations, diagnosis, and management as well as common complications seen in individuals with CF.
© American Academy of Pediatrics, 2021. All rights reserved.
Conflict of interest statement
AUTHOR DISCLOSUREDrs Dickinson and Collaco have disclosed no financial disclosures relevant to this article. This commentary does not contain a discussion of an unapproved/investigative use of commercial products/devices.
Figures
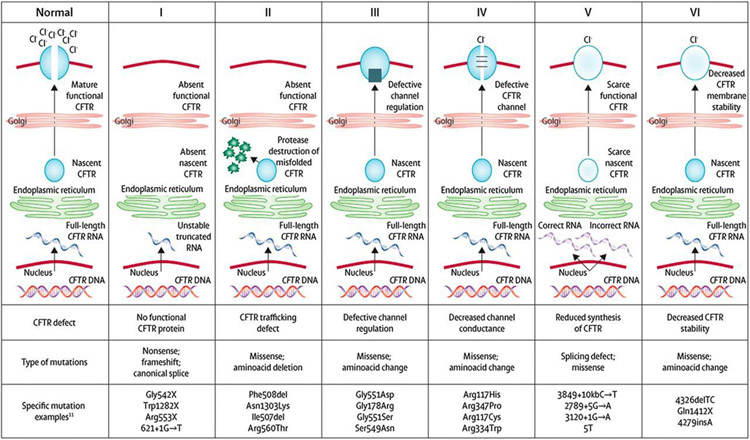

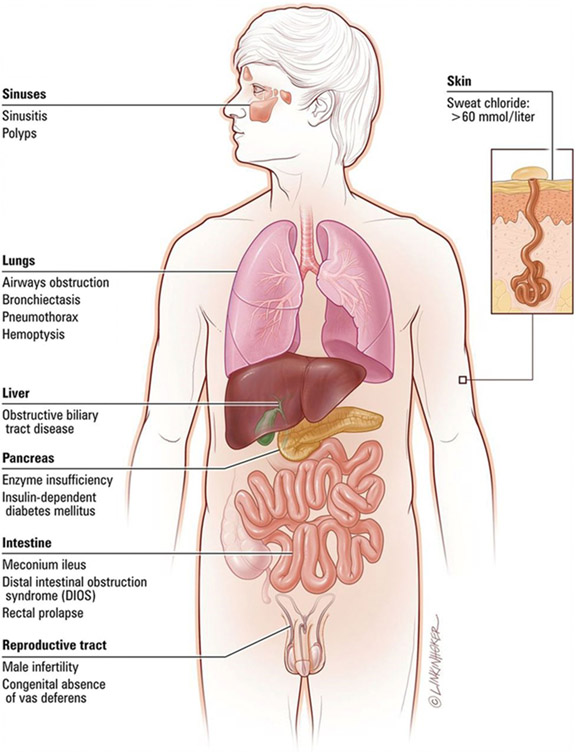
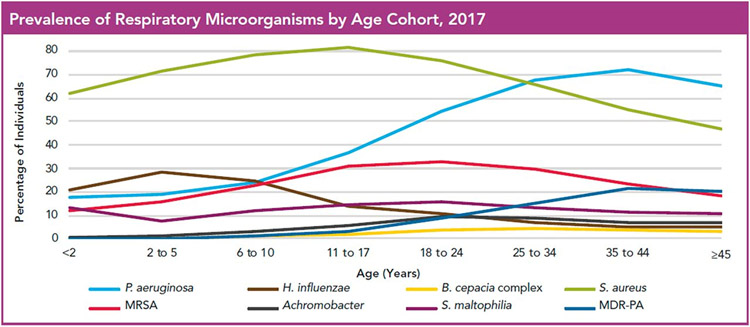
References
-
- Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease. Am J Dis Child. 1938;56(2):344–399
-
- Cystic Fibrosis Foundation Patient Registry. 2018 Patient Registry Annual Data Report. Bethesda, Maryland. Bethesda, Maryland.
-
- Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998;132(2):255–259 - PubMed
-
- Rohlfs EM, Zhou Z, Heim RA, et al. Cystic fibrosis carrier testing in an ethnically diverse US population. Clin Chem. 2011;57(6):841–848 - PubMed
-
- Carrier Testing for Cystic Fibrosis. Cystic Fibrosis Foundation.[internet] Https://www.cff.org/What-is-CF/Testing/Carrier-Testing-for-Cystic-Fibrosis/. Accessed August 1, 2019
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
