

Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm
- PMID: 33526886
- PMCID: PMC7961889
- DOI: 10.1038/s41592-020-01056-5
Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm
Abstract
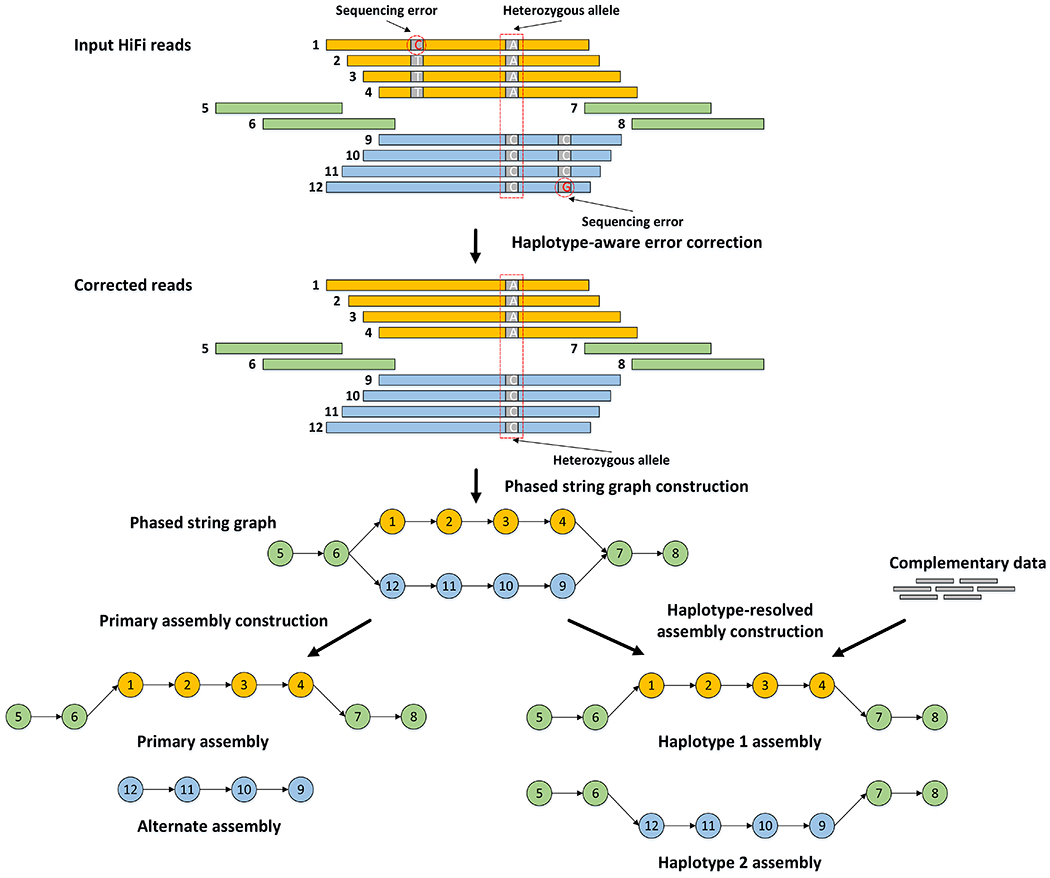
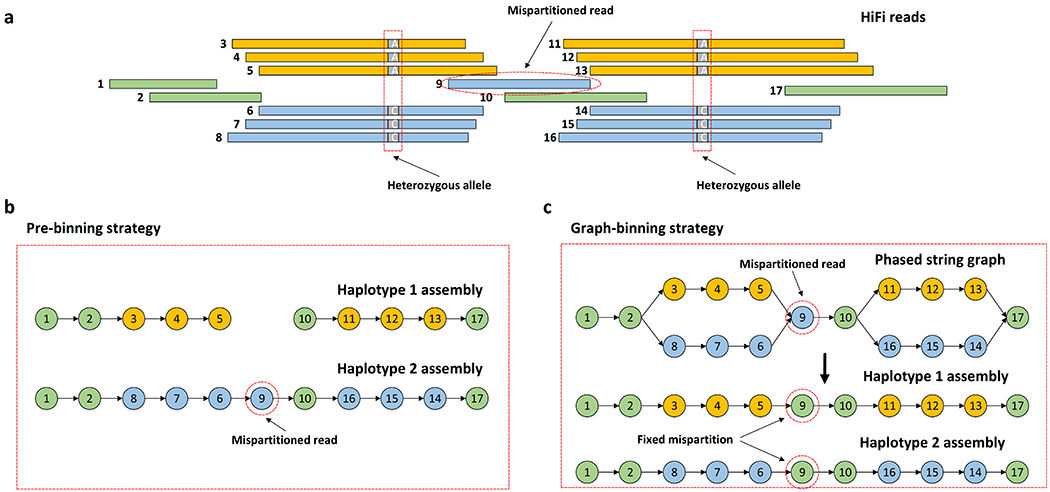
Haplotype-resolved de novo assembly is the ultimate solution to the study of sequence variations in a genome. However, existing algorithms either collapse heterozygous alleles into one consensus copy or fail to cleanly separate the haplotypes to produce high-quality phased assemblies. Here we describe hifiasm, a de novo assembler that takes advantage of long high-fidelity sequence reads to faithfully represent the haplotype information in a phased assembly graph. Unlike other graph-based assemblers that only aim to maintain the contiguity of one haplotype, hifiasm strives to preserve the contiguity of all haplotypes. This feature enables the development of a graph trio binning algorithm that greatly advances over standard trio binning. On three human and five nonhuman datasets, including California redwood with a ~30-Gb hexaploid genome, we show that hifiasm frequently delivers better assemblies than existing tools and consistently outperforms others on haplotype-resolved assembly.
Figures
References
-
- Chin C-S et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013). - PubMed
-
- Berlin K et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol 33, 623–630 (2015). - PubMed
-
- Kolmogorov M, Yuan J, Lin Y & Pevzner PA Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol 37, 540–546 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
