

Computation-guided optimization of split protein systems
- PMID: 33526893
- PMCID: PMC8084939
- DOI: 10.1038/s41589-020-00729-8
Computation-guided optimization of split protein systems
Abstract
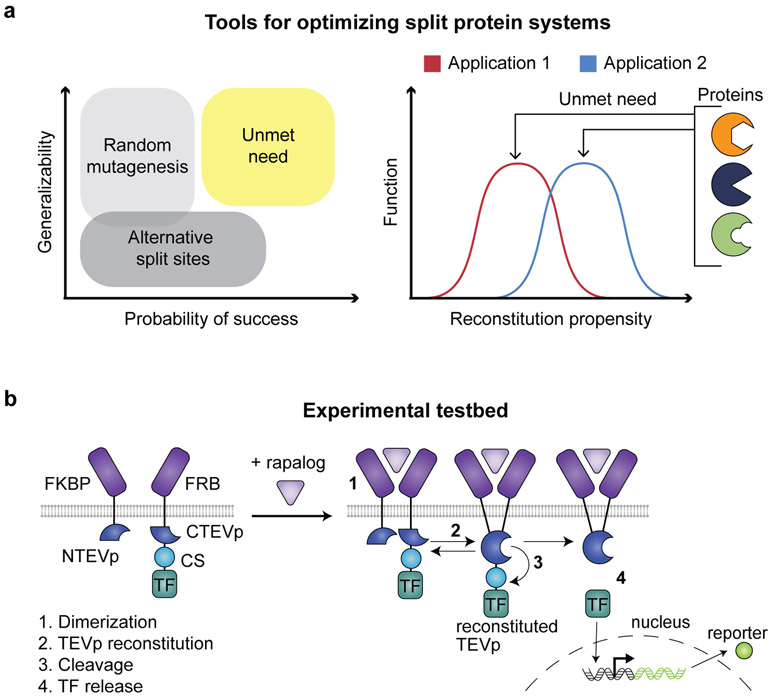
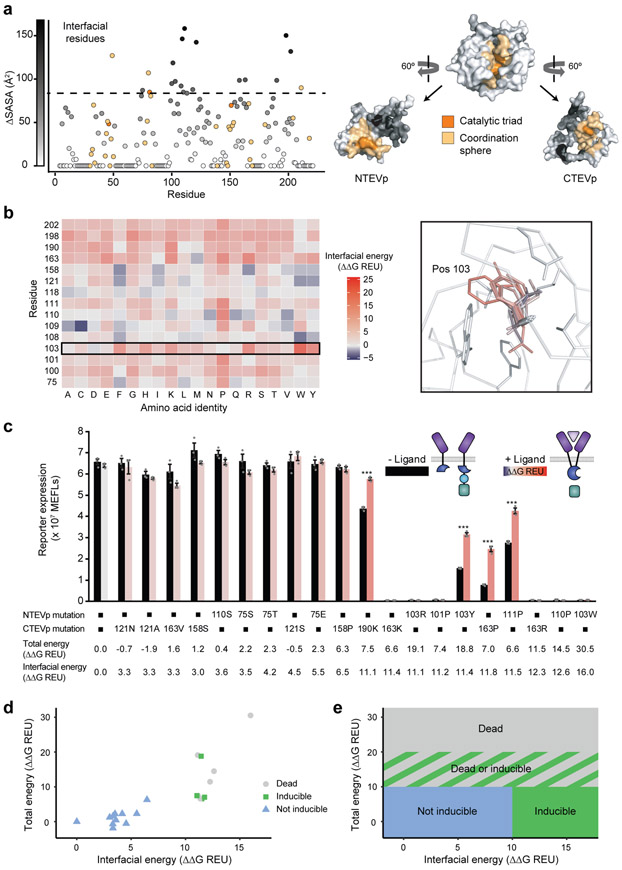
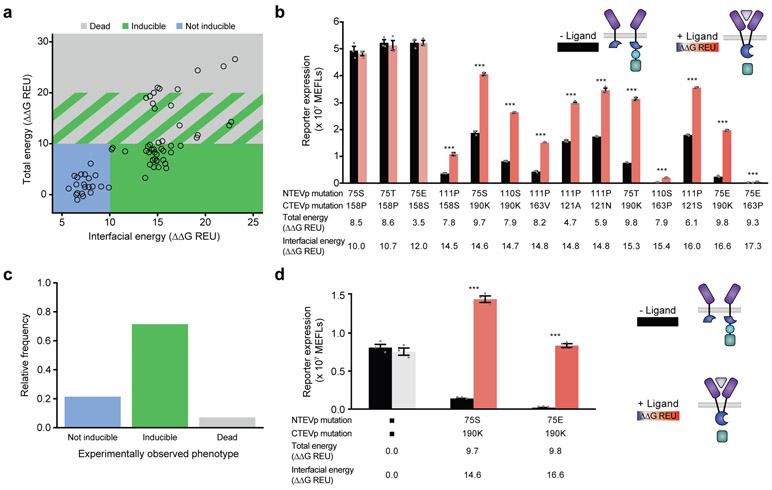
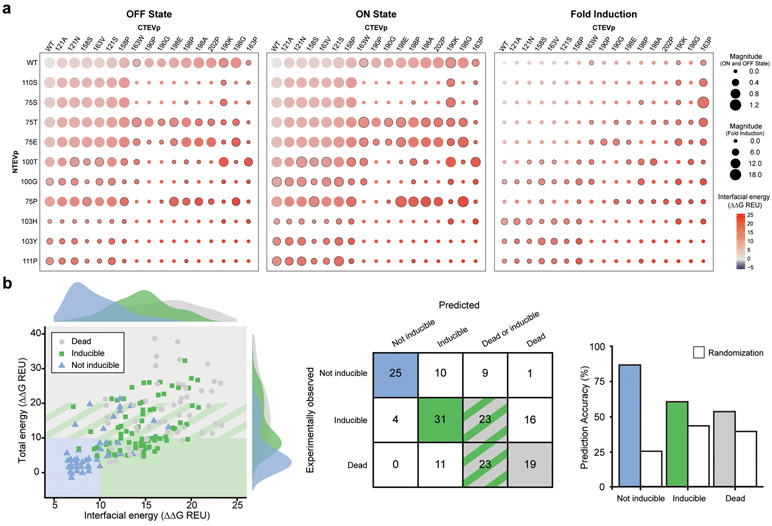
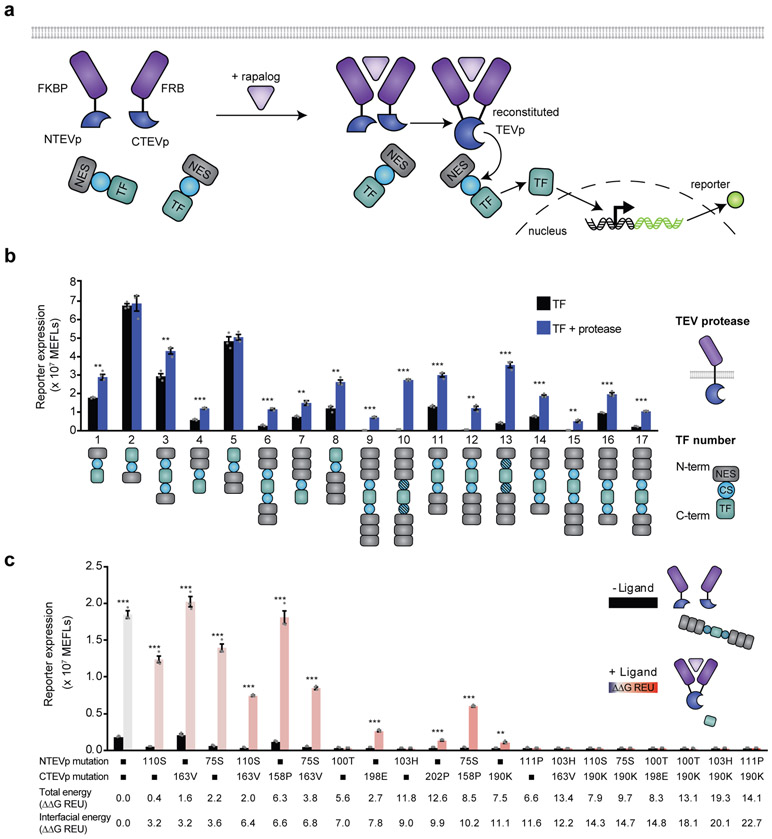
Splitting bioactive proteins into conditionally reconstituting fragments is a powerful strategy for building tools to study and control biological systems. However, split proteins often exhibit a high propensity to reconstitute, even without the conditional trigger, limiting their utility. Current approaches for tuning reconstitution propensity are laborious, context-specific or often ineffective. Here, we report a computational design strategy grounded in fundamental protein biophysics to guide experimental evaluation of a sparse set of mutants to identify an optimal functional window. We hypothesized that testing a limited set of mutants would direct subsequent mutagenesis efforts by predicting desirable mutant combinations from a vast mutational landscape. This strategy varies the degree of interfacial destabilization while preserving stability and catalytic activity. We validate our method by solving two distinct split protein design challenges, generating both design and mechanistic insights. This new technology will streamline the generation and use of split protein systems for diverse applications.
Figures
References
REFERENCES: METHODS-ONLY
-
- Eisenhaber F, Lijnzaad P, Argos P, Sander C & Scharf M The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. Journal of Computational Chemistry 16, 273–284 (1995).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
