

Genome-Scale Profiling Reveals Noncoding Loci Carry Higher Proportions of Concordant Data
- PMID: 33528497
- PMCID: PMC8136493
- DOI: 10.1093/molbev/msab026
Genome-Scale Profiling Reveals Noncoding Loci Carry Higher Proportions of Concordant Data
Abstract
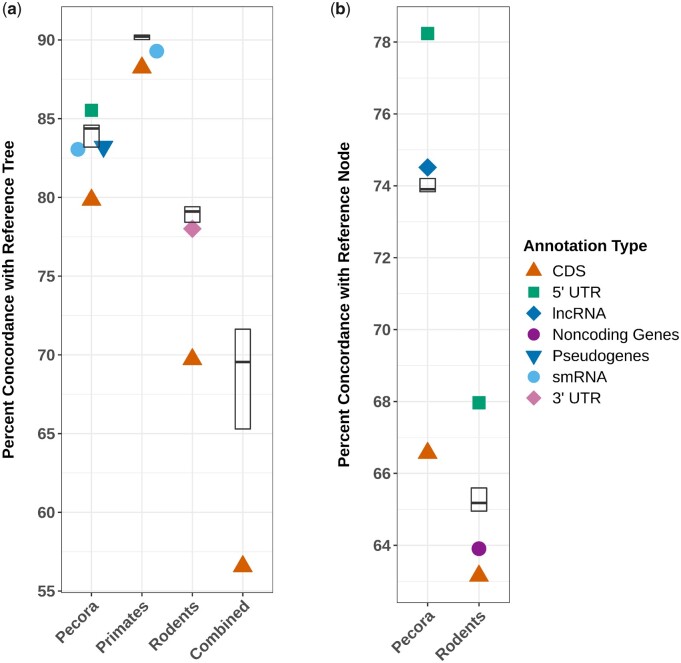
Many evolutionary relationships remain controversial despite whole-genome sequencing data. These controversies arise, in part, due to challenges associated with accurately modeling the complex phylogenetic signal coming from genomic regions experiencing distinct evolutionary forces. Here, we examine how different regions of the genome support or contradict well-established relationships among three mammal groups using millions of orthologous parsimony-informative biallelic sites (PIBS) distributed across primate, rodent, and Pecora genomes. We compared PIBS concordance percentages among locus types (e.g. coding sequences (CDS), introns, intergenic regions), and contrasted PIBS utility over evolutionary timescales. Sites derived from noncoding sequences provided more data and proportionally more concordant sites compared with those from CDS in all clades. CDS PIBS were also predominant drivers of tree incongruence in two cases of topological conflict. PIBS derived from most locus types provided surprisingly consistent support for splitting events spread across the timescales we examined, although we find evidence that CDS and intronic PIBS may, respectively and to a limited degree, inform disproportionately about older and younger splits. In this era of accessible wholegenome sequence data, these results:1) suggest benefits to more intentionally focusing on noncoding loci as robust data for tree inference and 2) reinforce the importance of accurate modeling, especially when using CDS data.
Keywords: bioinformatics; genomics; phylogenetics.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures


Similar articles
-
Larger, unfiltered datasets are more effective at resolving phylogenetic conflict: Introns, exons, and UCEs resolve ambiguities in Golden-backed frogs (Anura: Ranidae; genus Hylarana).Mol Phylogenet Evol. 2020 Oct;151:106899. doi: 10.1016/j.ympev.2020.106899. Epub 2020 Jun 24. Mol Phylogenet Evol. 2020. PMID: 32590046
-
Functions of noncoding sequences in mammalian genomes.Biochemistry (Mosc). 2014 Dec;79(13):1442-69. doi: 10.1134/S0006297914130021. Biochemistry (Mosc). 2014. PMID: 25749159 Review.
-
Phylogenomic Resolution of the Phylogeny of Laurasiatherian Mammals: Exploring Phylogenetic Signals within Coding and Noncoding Sequences.Genome Biol Evol. 2017 Aug 1;9(8):1998-2012. doi: 10.1093/gbe/evx147. Genome Biol Evol. 2017. PMID: 28830116 Free PMC article.
-
Identification and assessment of variable single-copy orthologous (SCO) nuclear loci for low-level phylogenomics: a case study in the genus Rosa (Rosaceae).BMC Evol Biol. 2019 Jul 24;19(1):152. doi: 10.1186/s12862-019-1479-z. BMC Evol Biol. 2019. PMID: 31340752 Free PMC article.
-
Mammalian phylogenomics comes of age.Trends Genet. 2004 Dec;20(12):631-9. doi: 10.1016/j.tig.2004.09.005. Trends Genet. 2004. PMID: 15522459 Review.
Cited by
-
A genomic timescale for placental mammal evolution.Science. 2023 Apr 28;380(6643):eabl8189. doi: 10.1126/science.abl8189. Epub 2023 Apr 28. Science. 2023. PMID: 37104581 Free PMC article.
-
Reference-free discovery of nuclear SNPs permits accurate, sensitive identification of Carya (hickory) species and hybrids.Appl Plant Sci. 2022 Jan 20;10(1):e11455. doi: 10.1002/aps3.11455. eCollection 2022 Jan-Feb. Appl Plant Sci. 2022. PMID: 35228913 Free PMC article.
-
Accurate, scalable, and fully automated inference of species trees from raw genome assemblies using ROADIES.Proc Natl Acad Sci U S A. 2025 May 13;122(19):e2500553122. doi: 10.1073/pnas.2500553122. Epub 2025 May 2. Proc Natl Acad Sci U S A. 2025. PMID: 40314967 Free PMC article.
-
The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order.Genes (Basel). 2023 Jul 1;14(7):1387. doi: 10.3390/genes14071387. Genes (Basel). 2023. PMID: 37510292 Free PMC article. Review.
-
Plastid phylogenomics and cytonuclear discordance in Rubioideae, Rubiaceae.PLoS One. 2024 May 20;19(5):e0302365. doi: 10.1371/journal.pone.0302365. eCollection 2024. PLoS One. 2024. PMID: 38768140 Free PMC article.
References
-
- Aguileta G, Marthey S, Chiapello H, Lebrun M-H, Rodolphe F, Fournier E, Gendrault-Jacquemard A, Giraud T.. 2008. Assessing the performance of single-copy genes for recovering robust phylogenies. Syst Biol 57(4):613–627. - PubMed
-
- Bejerano G. 2004. Ultraconservedelements in the human genome. Science 304(5675):1321–1325. - PubMed
-
- Bleidorn C. 2017. Sources of error and incongruence in phylogenomic analyses. Phylogenomics 173–193, doi:10.1007/978-3-319-54064-1_9
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
