

Bite of the wolf: innate immune responses propagate autoimmunity in lupus
- PMID: 33529160
- PMCID: PMC7843222
- DOI: 10.1172/JCI144918
Bite of the wolf: innate immune responses propagate autoimmunity in lupus
Abstract
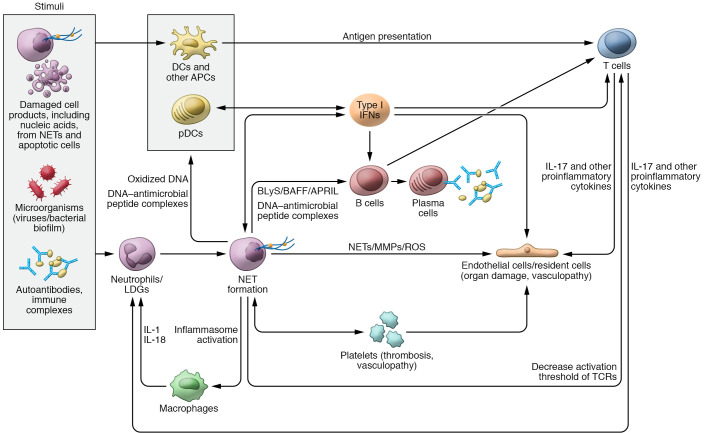
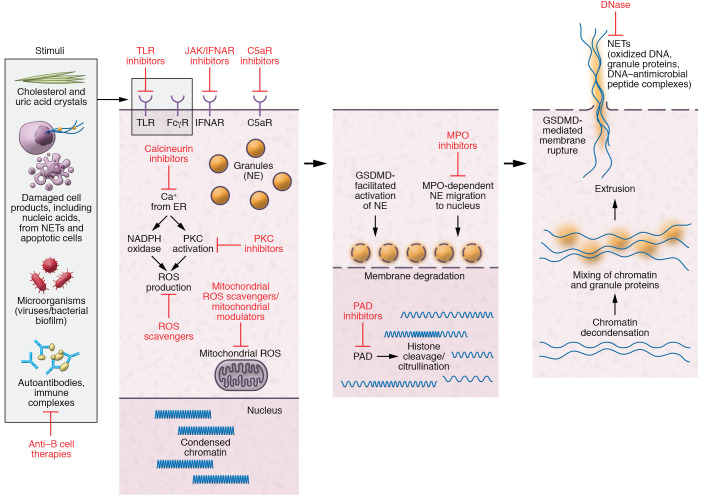
The etiopathogenesis of systemic lupus erythematosus (SLE), a clinically heterogeneous multisystemic syndrome that derives its name from the initial characterization of facial lesions that resemble the bite of a wolf, is considered a complex, multifactorial interplay between underlying genetic susceptibility factors and the environment. Prominent pathogenic factors include the induction of aberrant cell death pathways coupled with defective cell death clearance mechanisms that promote excessive externalization of modified cellular and nuclear debris with subsequent loss of tolerance to a wide variety of autoantigens and innate and adaptive immune dysregulation. While abnormalities in adaptive immunity are well recognized and are key to the pathogenesis of SLE, recent findings have emphasized fundamental roles of the innate immune system in the initiation and propagation of autoimmunity and the development of organ damage in this disease. This Review focuses on recent discoveries regarding the role of components of the innate immune system, specifically neutrophils and interferons, in promoting various aspects of lupus pathogenesis, with potential implications for novel therapeutic strategies.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
