

Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents
- PMID: 33530139
- PMCID: PMC7837145
- DOI: 10.1186/s40959-016-0010-6
Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents
Abstract
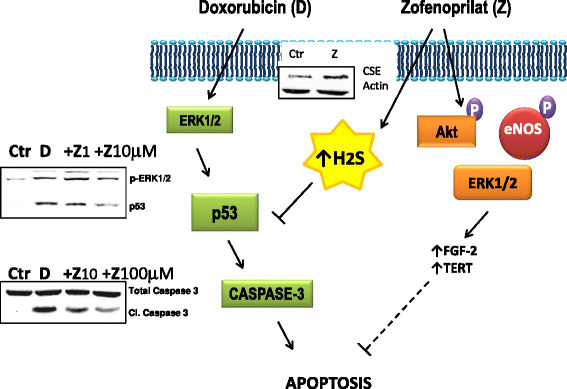
The vascular endothelium plays a fundamental role in the maintenance of tissue homeostasis, regulating local blood flow and other physiological processes. Chemotherapeutic drugs and target therapies, including antiangiogenic drugs targeting vascular endothelial growth factor (VEGF) or its receptors, not only efficiently act against tumor growth, but may also induce endothelial dysfunction and cardiovascular toxicity. Continued research efforts aim to better understand, prevent and mitigate these chemotherapy associated cardiovascular diseases. Conventional chemotherapeutic agents, such as anthracyclines, platinum compounds, and taxanes, and newer targeted agents, such as bevacizumab, trastuzumab, and tyrosine kinase inhibitors, have known risk of cardiovascular toxicity, which can limit their effectiveness by promoting increased morbidity and/or mortality. This review describes a) the activity of anticancer agents in inducing endothelial dysfunction, b) the metabolic pathways and signalling cascades which may be targeted by protective agents able to maintain or restore endothelial cell function, such as endothelial nitric oxide synthase/fibroblast growth factor-2 (eNOS-FGF-2) pathway, and c) the drugs/strategies reported to improve endothelial function and to reduce the risks of cardiovascular diseases such as angiotensin converting enzyme inhibitors (ACEi) and beta blockers, that are fundamental therapies in chronic heart failure (HF), as well as non-standard HF treatments such ad nitric oxide donors and antioxidant strategies. There is increasing interest in whether ACEi, beta-blockers, and/or statins might prevent and/or therapeutically control cardiotoxic effects in cancer patients. Maintaining endothelial function during or following treatments with chemotherapeutic agents, without affecting anti-tumor drug-effectiveness, is essential for preserving or recovering cardiovascular homeostasis. In this respect, the early detection and immediate therapy of cardiovascular toxicity appear crucial for substantial recovery of cardiac function in cancer patients.
Keywords: Nitric Oxide; Sorafenib; Sunitinib; Trastuzumab; Vascular Endothelial Growth Factor.
Figures



References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous