

Hypoxia-Induced S100A8 Expression Activates Microglial Inflammation and Promotes Neuronal Apoptosis
- PMID: 33530496
- PMCID: PMC7866104
- DOI: 10.3390/ijms22031205
Hypoxia-Induced S100A8 Expression Activates Microglial Inflammation and Promotes Neuronal Apoptosis
Abstract
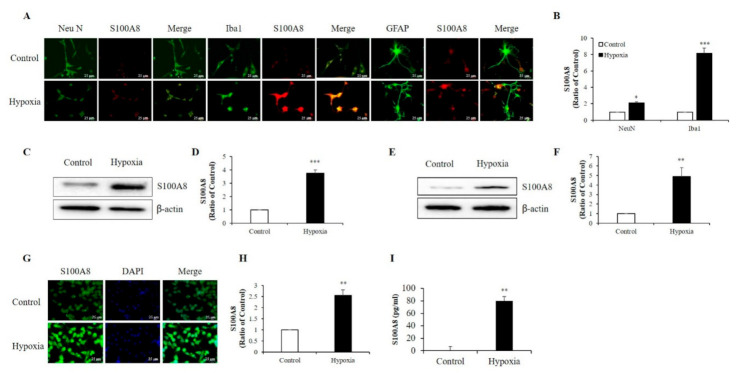
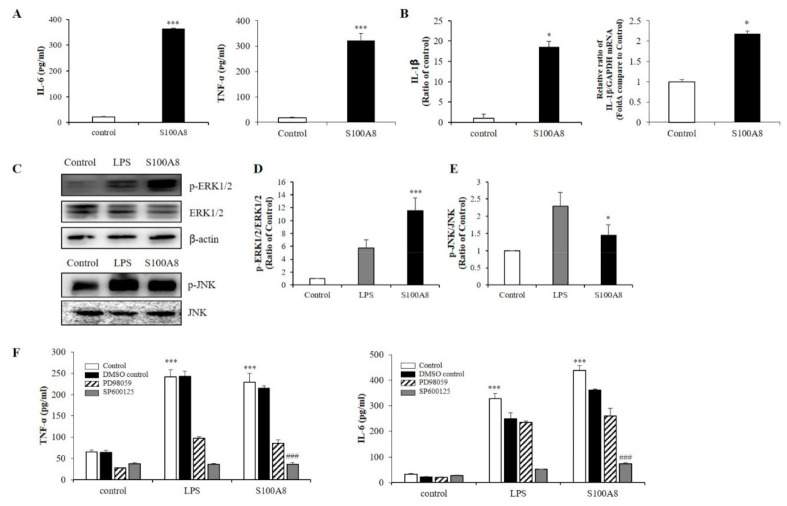
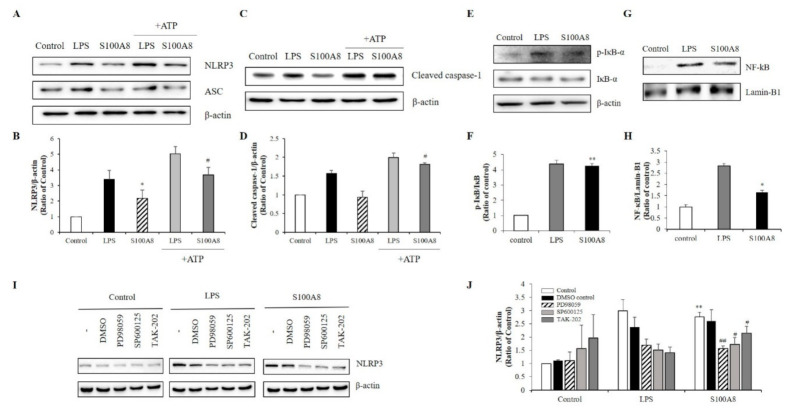
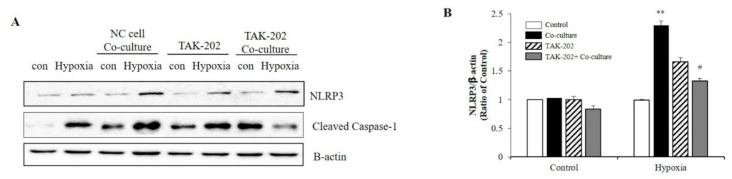
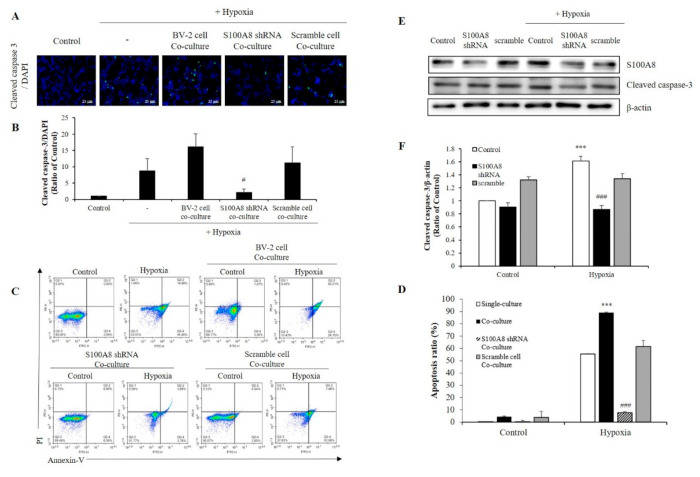
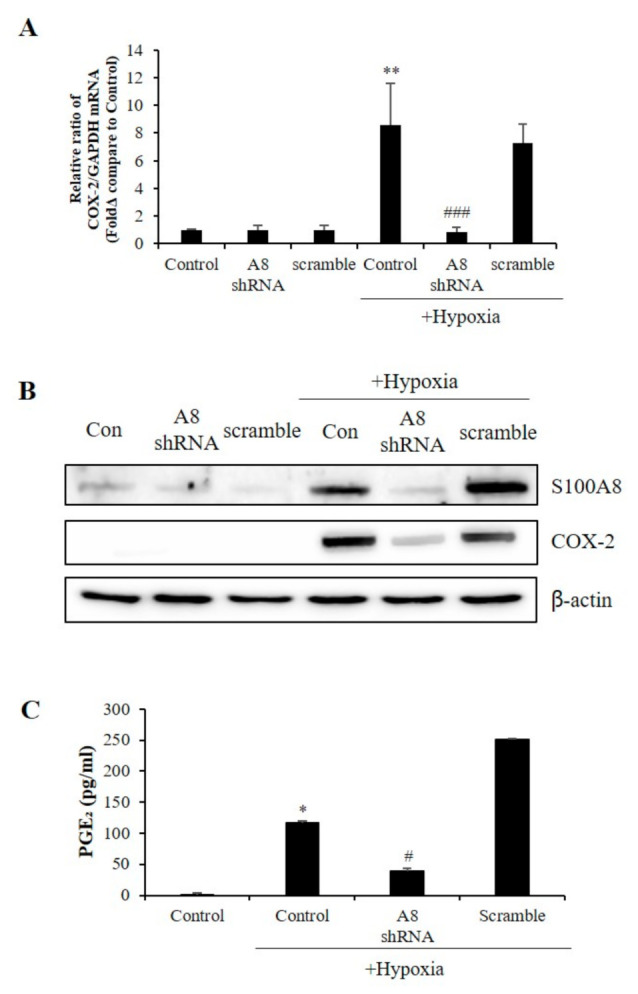
S100 calcium-binding protein A8 (S100A8), a danger-associated molecular pattern, has emerged as an important mediator of the pro-inflammatory response. Some S100 proteins play a prominent role in neuroinflammatory disorders and increase the secretion of pro-inflammatory cytokines in microglial cells. The aim of this study was to determine whether S100A8 induced neuronal apoptosis during cerebral hypoxia and elucidate its mechanism of action. In this study, we reported that the S100A8 protein expression was increased in mouse neuronal and microglial cells when exposed to hypoxia, and induced neuroinflammation and neuronal apoptosis. S100A8, secreted from neurons under hypoxia, activated the secretion of tumor necrosis factor (TNF-α) and interleukin-6 (IL-6) through phosphorylation of extracellular-signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in microglia. Also, phosphorylation of ERK via the TLR4 receptor induced the priming of the NLRP3 inflammasome. The changes in Cyclooxygenase-2 (COX-2) expression, a well-known inflammatory activator, were regulated by the S100A8 expression in microglial cells. Knockdown of S100A8 levels by using shRNA revealed that microglial S100A8 expression activated COX-2 expression, leading to neuronal apoptosis under hypoxia. These results suggested that S100A8 may be an important molecule for bidirectional microglia-neuron communication and a new therapeutic target for neurological disorders caused by microglial inflammation during hypoxia.
Keywords: COX-2; S100A8; hypoxia; inflammasome; microglia; neuronal apoptosis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
