

Biallelic loss-of-function in NRAP is a cause of recessive dilated cardiomyopathy
- PMID: 33534821
- PMCID: PMC7857588
- DOI: 10.1371/journal.pone.0245681
Biallelic loss-of-function in NRAP is a cause of recessive dilated cardiomyopathy
Abstract
Background: Familial dilated cardiomyopathy (DCM) is typically a monogenic disorder with dominant inheritance. Although over 40 genes have been linked to DCM, more than half of the patients undergoing comprehensive genetic testing are left without molecular diagnosis. Recently, biallelic protein-truncating variants (PTVs) in the nebulin-related anchoring protein gene (NRAP) were identified in a few patients with sporadic DCM.
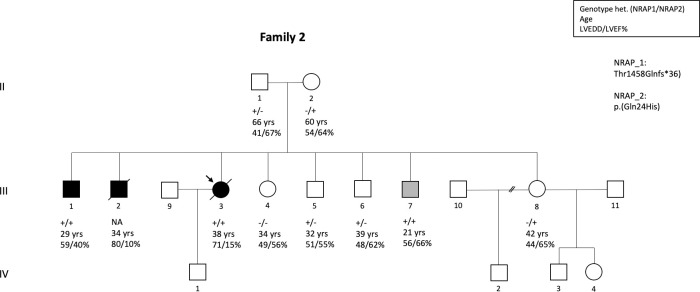
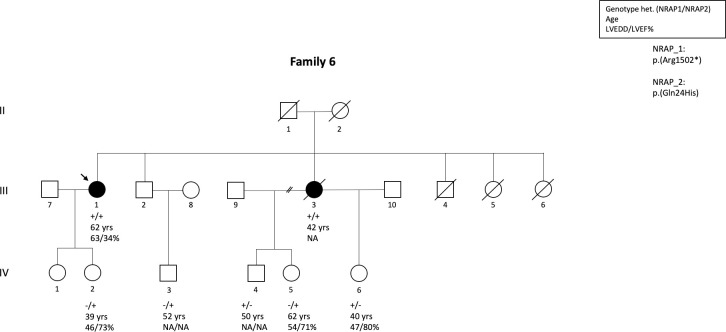
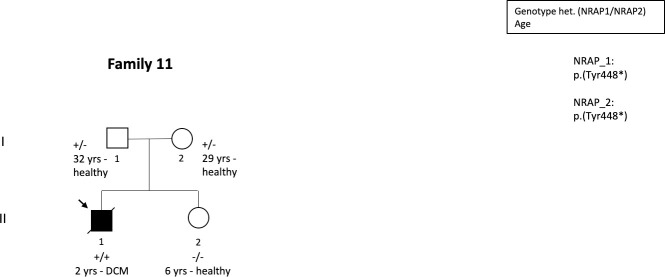
Methods and results: We determined the frequency of rare NRAP variants in a cohort of DCM patients and control patients to further evaluate role of this gene in cardiomyopathies. A retrospective analysis of our internal variant database consisting of 31,639 individuals who underwent genetic testing (either panel or direct exome sequencing) was performed. The DCM group included 577 patients with either a confirmed or suspected DCM diagnosis. A control cohort of 31,062 individuals, including 25,912 individuals with non-cardiac (control group) and 5,150 with non-DCM cardiac indications (Non-DCM cardiac group). Biallelic (n = 6) or two (n = 5) NRAP variants (two PTVs or PTV+missense) were identified in 11 unrelated probands with DCM (1.9%) but none of the controls. None of the 11 probands had an alternative molecular diagnosis. Family member testing supports co-segregation. Biallelic or potentially biallelic NRAP variants were enriched in DCM vs. controls (OR 1052, p<0.0001). Based on the frequency of NRAP PTVs in the gnomAD reference population, and predicting full penetrance, biallelic NRAP variants could explain 0.25%-2.46% of all DCM cases.
Conclusion: Loss-of-function in NRAP is a cause for autosomal recessive dilated cardiomyopathy, supporting its inclusion in comprehensive genetic testing.
Conflict of interest statement
Drs. Koskenvuo, Saarinen, Ahonen, Tommiska, Seppälä, Tuupanen, Kangas-Kontio, Schleit, Hathaway, Kytölä, Muona, Sistonen, Salmenperä, Gentile, Paananen, Myllykangas, Alastalo are full-time employees of Blueprint Genetics, a Quest Diagnostics Company, which offers genetic diagnostics for cardiomyopathies. The funder provided support in the form of salaries for authors [JWK, IS, SA, JT, ST, TKK, JS, JH, VK, MM, JS, PS, MG, JP, SM, TPA], but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the ‘author contributions’ section. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Figures
References
-
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29: 270–276. 10.1093/eurheartj/ehm342 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
