

Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders
- PMID: 33535037
- PMCID: PMC7903328
- DOI: 10.1016/j.celrep.2021.108718
Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders
Abstract
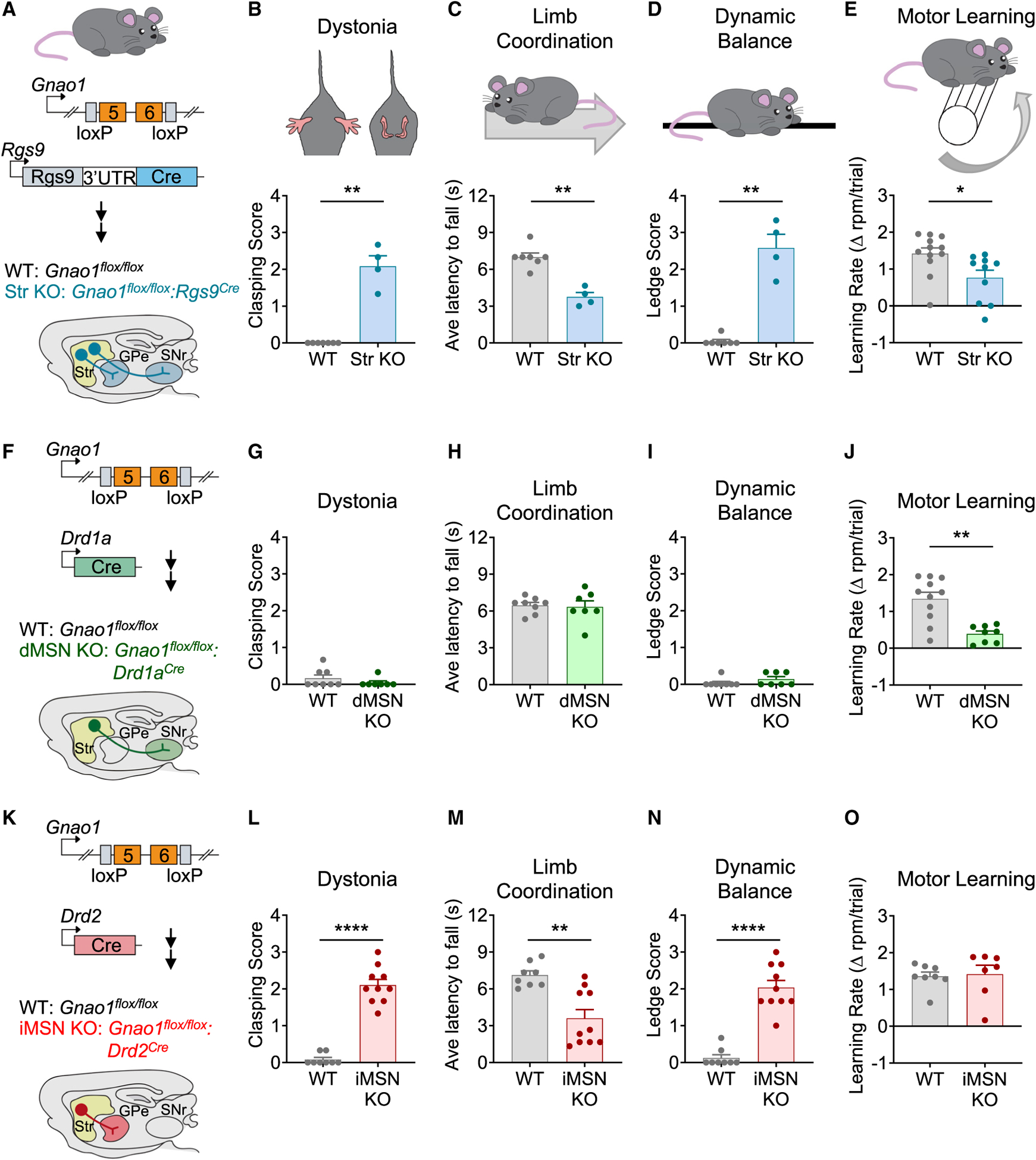
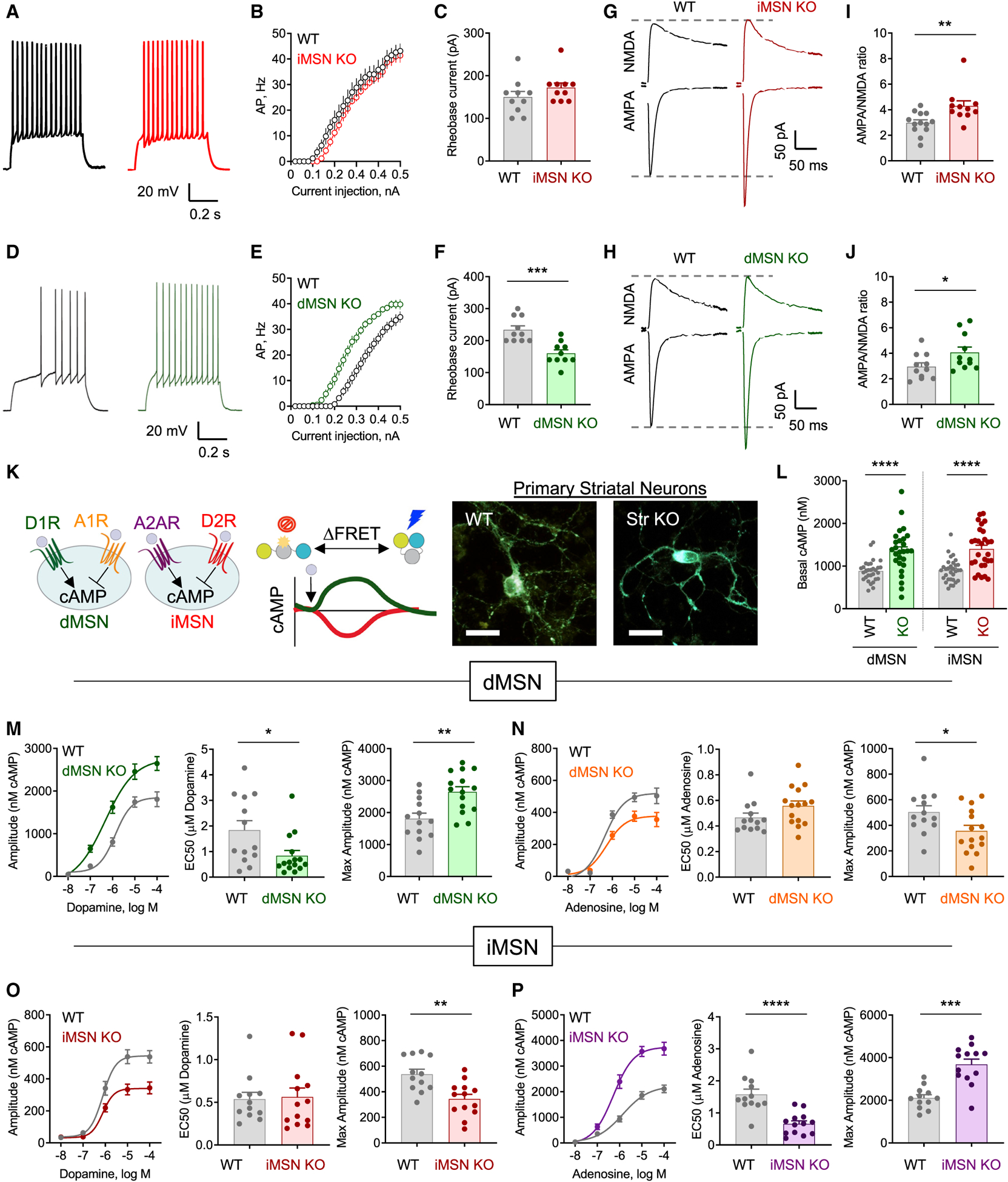
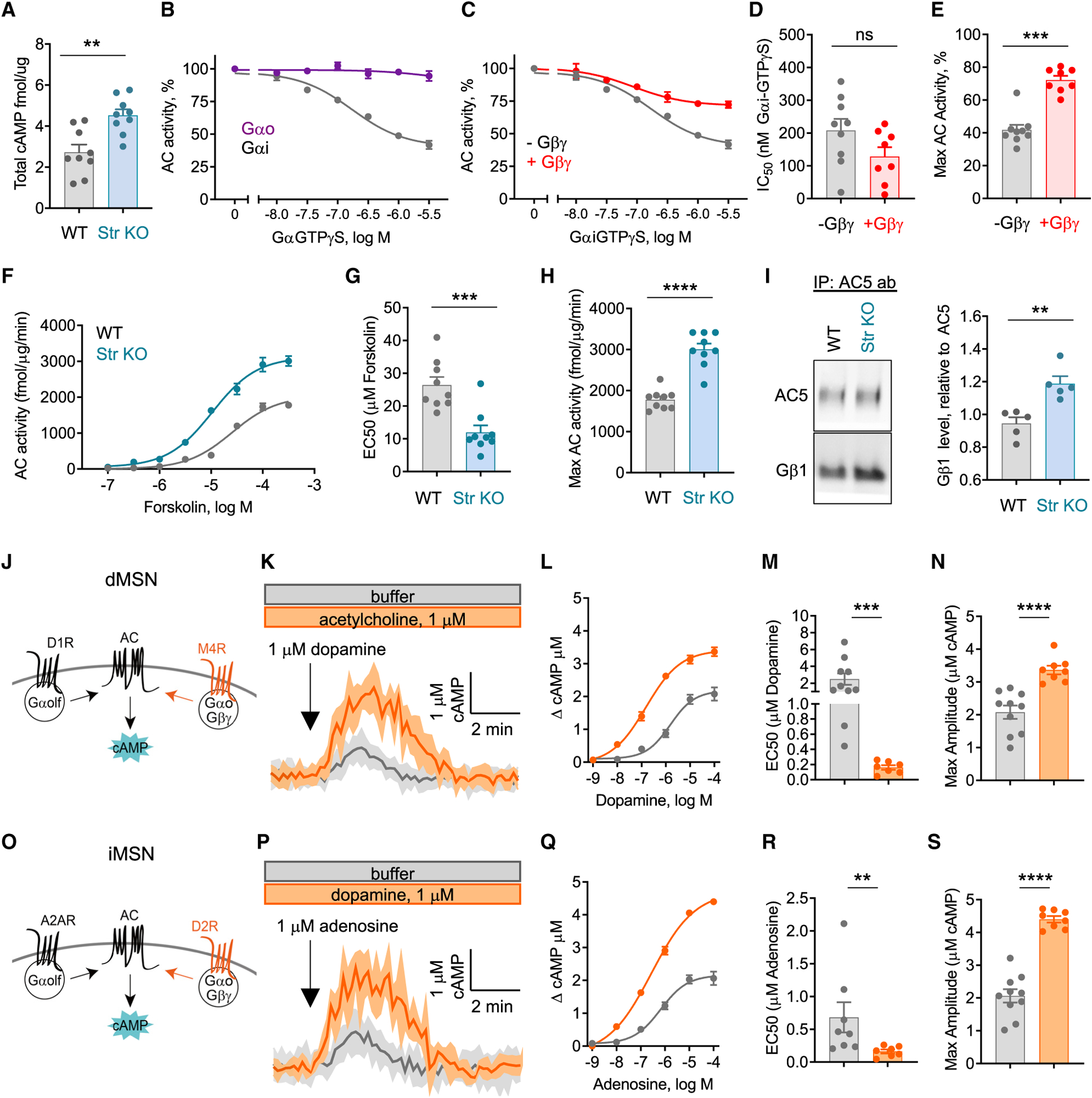
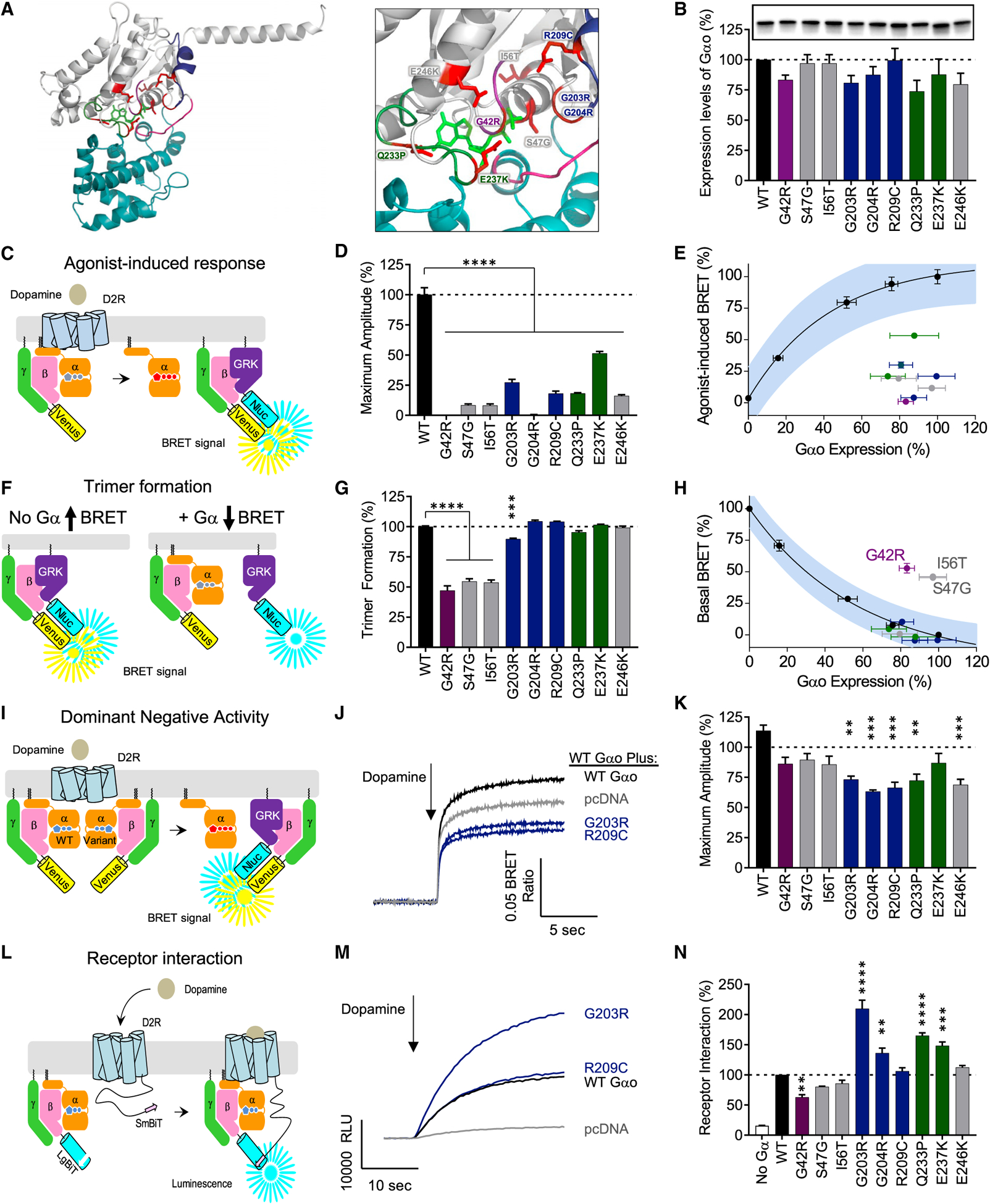
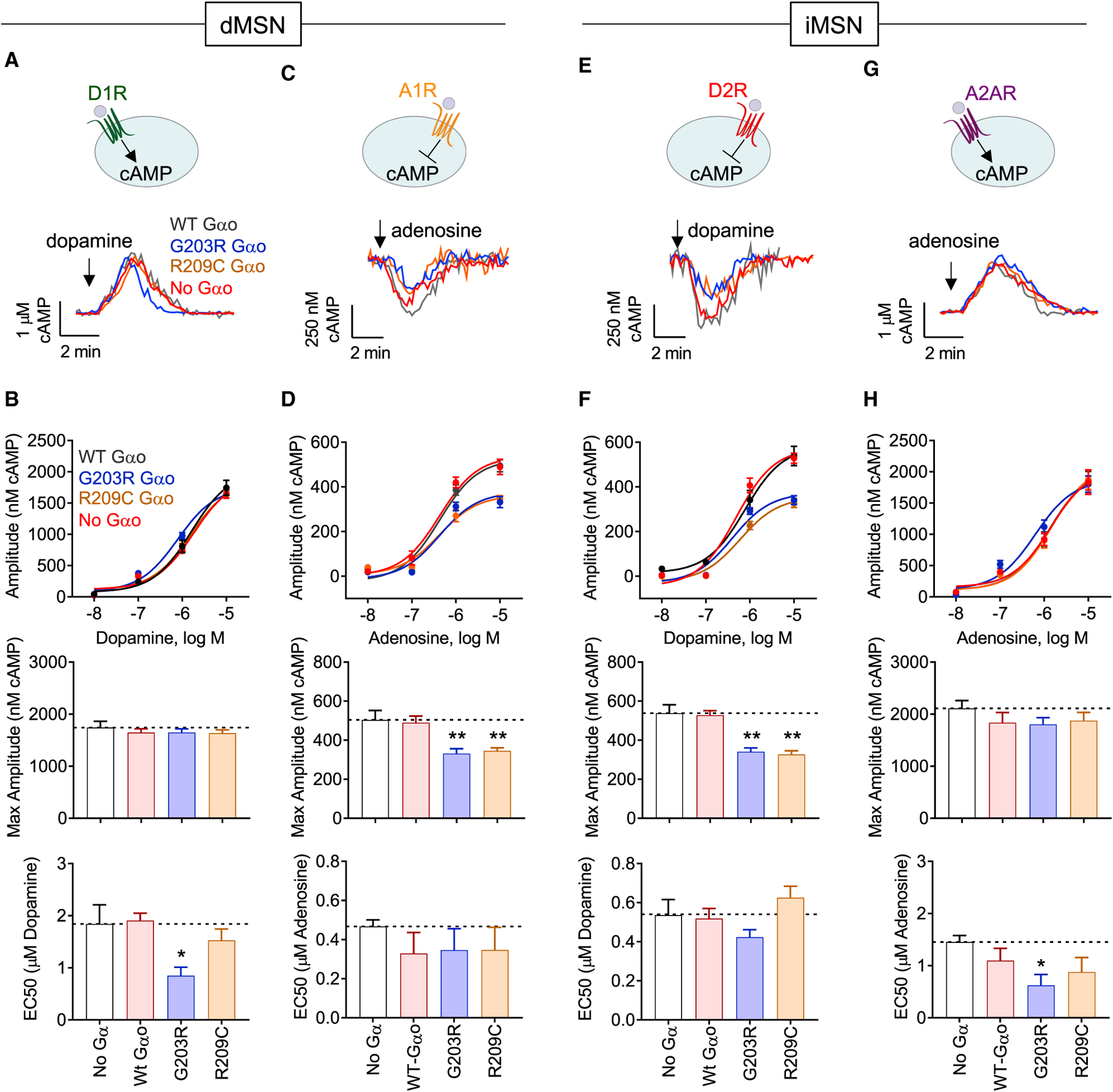
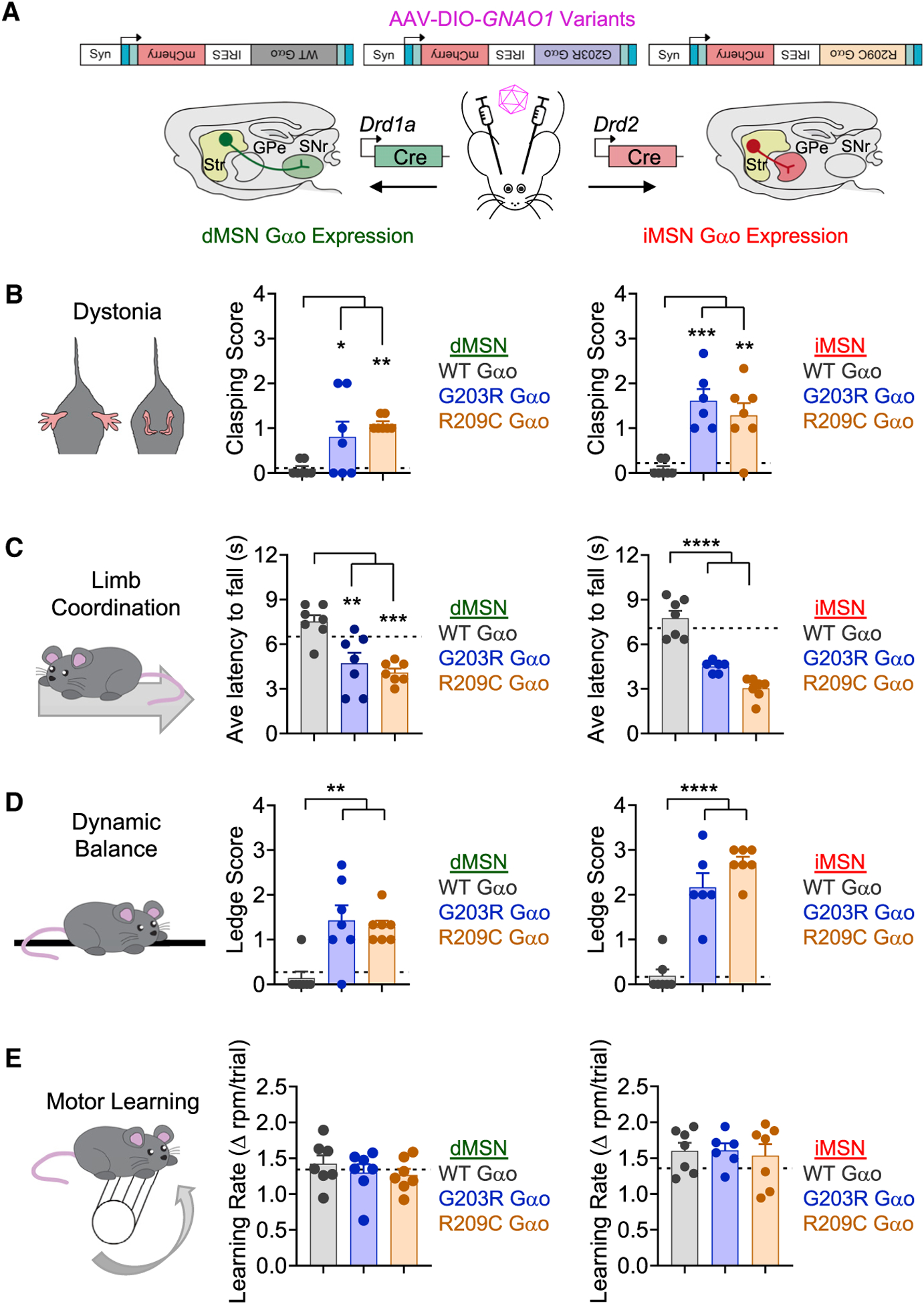
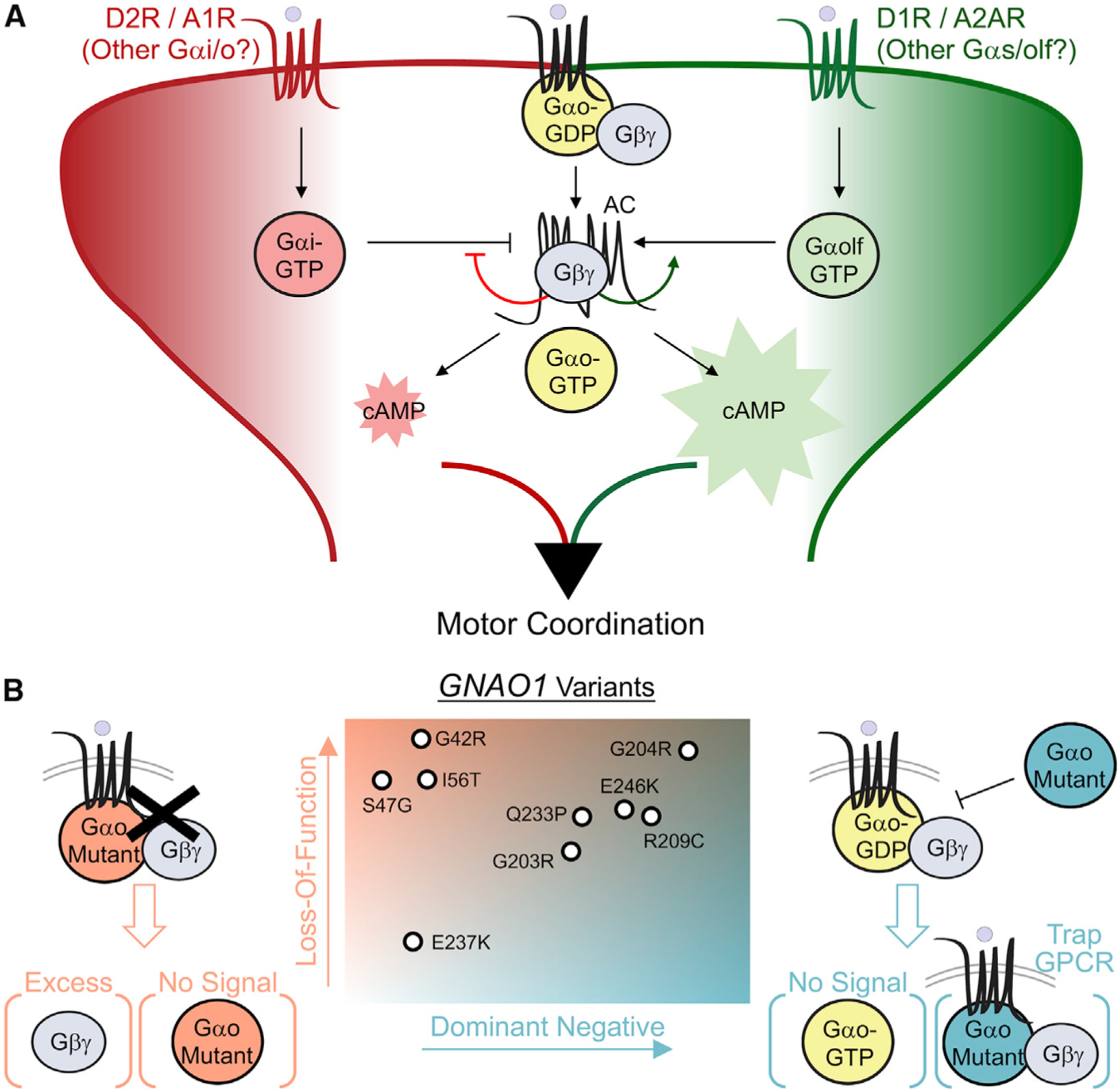
The G protein alpha subunit o (Gαo) is one of the most abundant proteins in the nervous system, and pathogenic mutations in its gene (GNAO1) cause movement disorder. However, the function of Gαo is ill defined mechanistically. Here, we show that Gαo dictates neuromodulatory responsiveness of striatal neurons and is required for movement control. Using in vivo optical sensors and enzymatic assays, we determine that Gαo provides a separate transduction channel that modulates coupling of both inhibitory and stimulatory dopamine receptors to the cyclic AMP (cAMP)-generating enzyme adenylyl cyclase. Through a combination of cell-based assays and rodent models, we demonstrate that GNAO1-associated mutations alter Gαo function in a neuron-type-specific fashion via a combination of a dominant-negative and loss-of-function mechanisms. Overall, our findings suggest that Gαo and its pathological variants function in specific circuits to regulate neuromodulatory signals essential for executing motor programs.
Keywords: GPCR; Gαo; cAMP; disease mechanisms; heterotrimeric G proteins; movement disorder; mutations; neuromodulation; striatum; synaptic plasticity.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Ananth AL, Robichaux-Viehoever A, Kim YM, Hanson-Kahn A, Cox R, Enns GM, Strober J, Willing M, Schlaggar BL, Wu YW, and Bernstein JA (2016). Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr. Neurol 59, 81–84. - PubMed
-
- Arshavsky VY, Dumke CL, Zhu Y, Artemyev NO, Skiba NP, Hamm HE, and Bownds MD (1994). Regulation of transducin GTPase activity in bovine rod outer segments. J. Biol. Chem 269, 19882–19887. - PubMed
-
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, and Borrelli E (1995). Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature 377, 424–428. - PubMed
-
- Berardelli A, Rothwell JC, Hallett M, Thompson PD, Manfredi M, and Marsden CD (1998). The pathophysiology of primary dystonia. Brain 121, 1195–1212. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
