

Hermansky-Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease
- PMID: 33536261
- PMCID: PMC9488956
- DOI: 10.1183/16000617.0193-2020
Hermansky-Pudlak syndrome pulmonary fibrosis: a rare inherited interstitial lung disease
Abstract
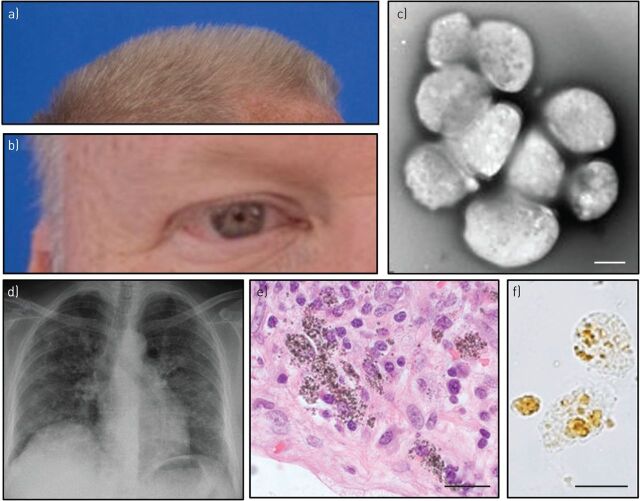
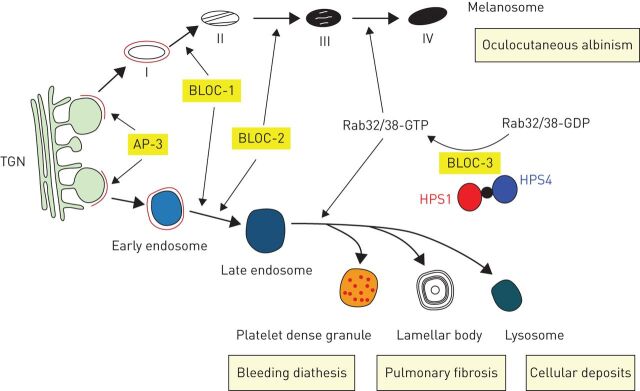
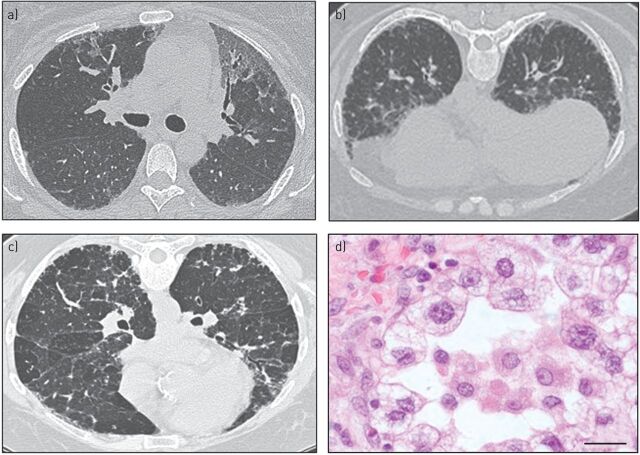
Pulmonary fibrosis is a progressive interstitial lung disease of unknown aetiology with a poor prognosis. Studying genetic diseases associated with pulmonary fibrosis provides insights into the pathogenesis of the disease. Hermansky-Pudlak syndrome (HPS), a rare autosomal recessive disorder characterised by abnormal biogenesis of lysosome-related organelles, manifests with oculocutaneous albinism and excessive bleeding of variable severity. Pulmonary fibrosis is highly prevalent in three out of 10 genetic types of HPS (HPS-1, HPS-2 and HPS-4). Thus, genotyping of individuals with HPS is clinically relevant. HPS-1 tends to affect Puerto Rican individuals due to a genetic founder effect. HPS pulmonary fibrosis shares some clinical features with idiopathic pulmonary fibrosis (IPF), including dyspnoea, cough, restrictive lung physiology and computed tomography (CT) findings of fibrosis. In contrast to IPF, HPS pulmonary fibrosis generally affects children (HPS-2) or middle-aged adults (HPS-1 or HPS-4) and may be associated with ground-glass opacification in CT scans. Histopathology of HPS pulmonary fibrosis, and not IPF, shows vacuolated hyperplastic type II cells with enlarged lamellar bodies and alveolar macrophages with lipofuscin-like deposits. Antifibrotic drugs approved as treatment for IPF are not approved for HPS pulmonary fibrosis. However, lung transplantation has been performed in patients with severe HPS pulmonary fibrosis. HPS pulmonary fibrosis serves as a model for studying fibrotic lung disease and fibrosis in general.
The content of this work is not subject to copyright. Design and branding are copyright ©ERS 2021.
Conflict of interest statement
Conflict of interest: T. Yokoyama has nothing to disclose. Conflict of interest: B.R. Gochuico has nothing to disclose.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical