

SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors
- PMID: 33536335
- PMCID: PMC8017720
- DOI: 10.1073/pnas.2015654118
SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors
Abstract
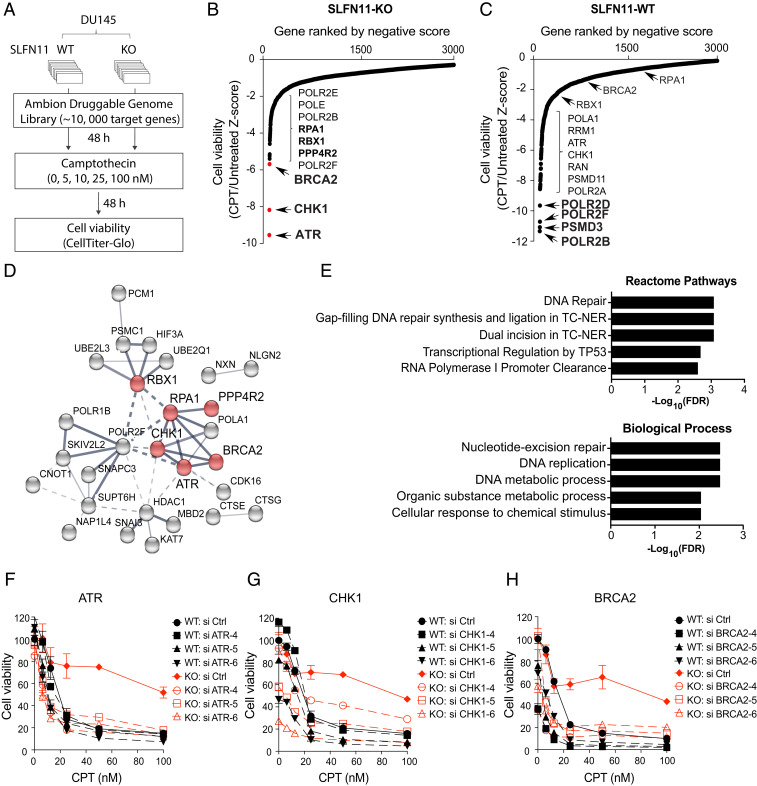
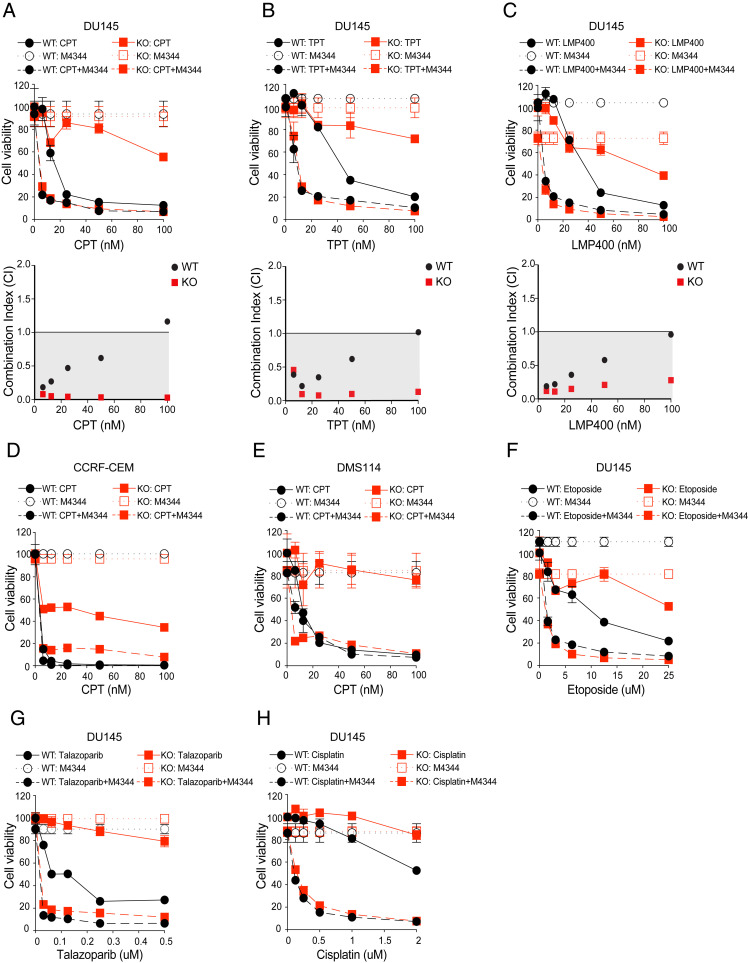
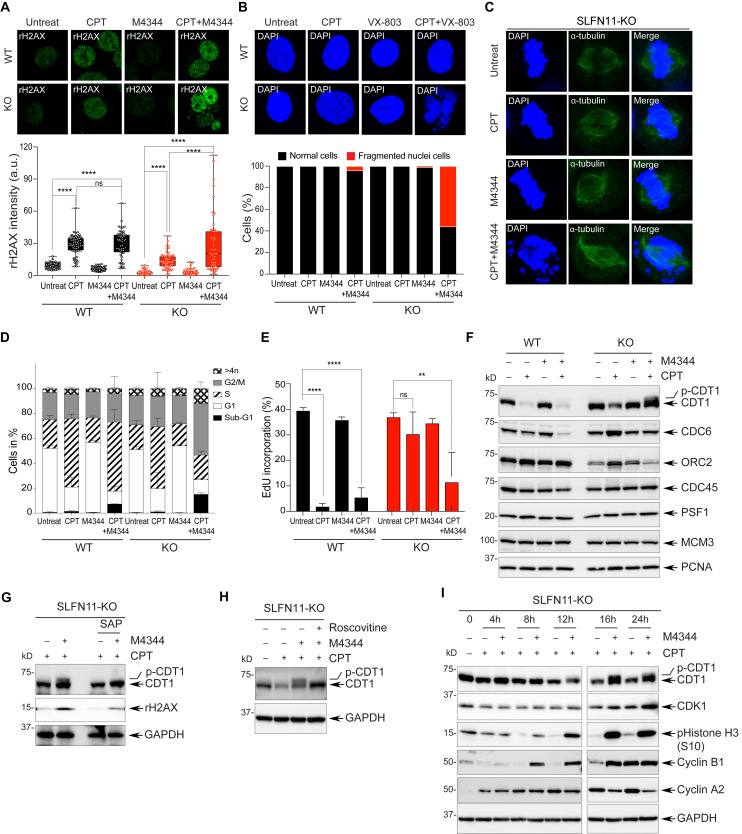
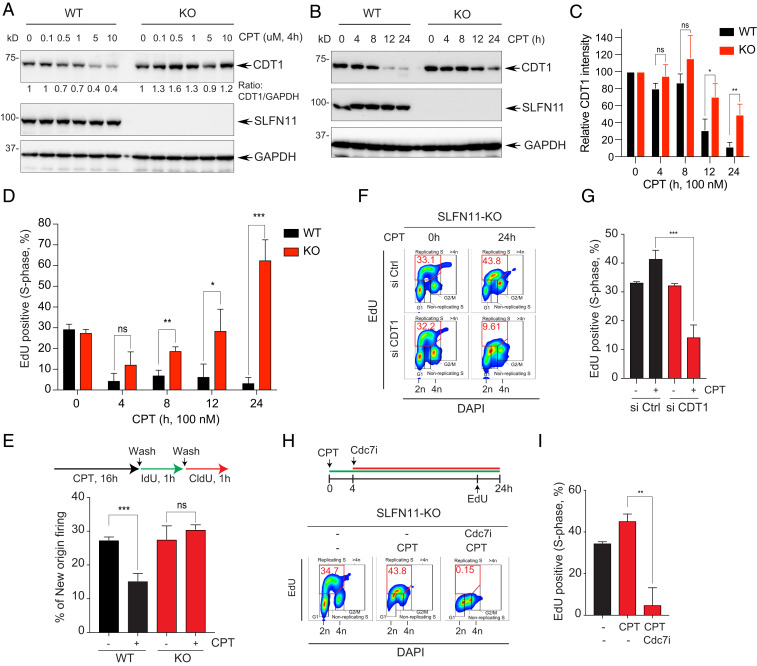
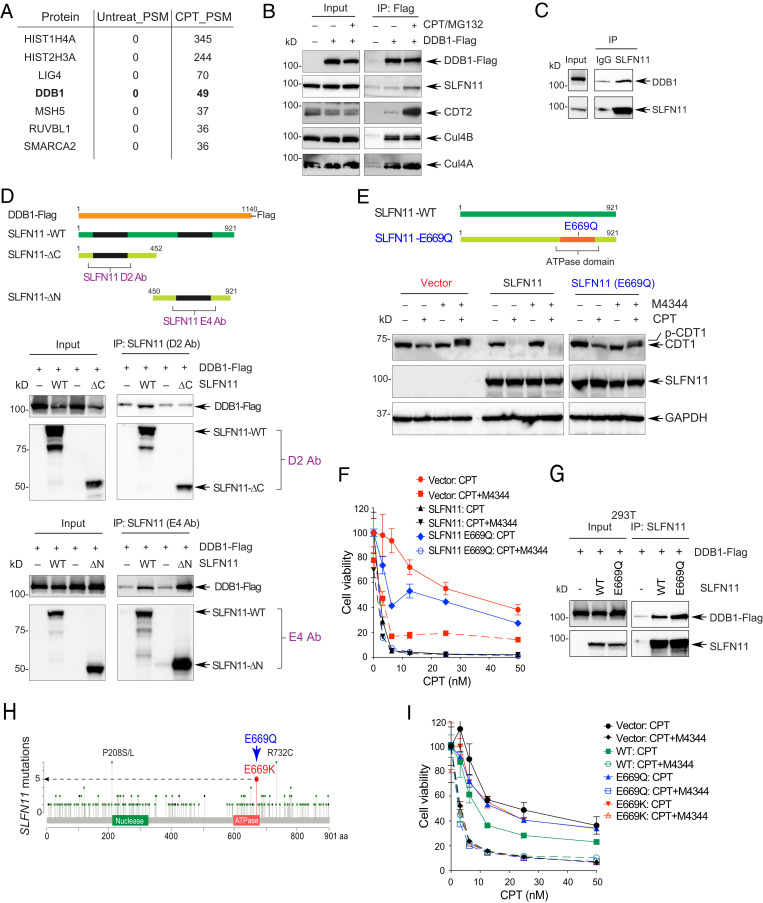
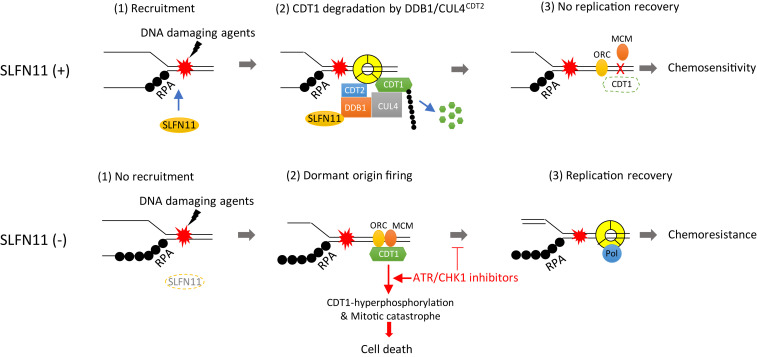
Schlafen-11 (SLFN11) inactivation in ∼50% of cancer cells confers broad chemoresistance. To identify therapeutic targets and underlying molecular mechanisms for overcoming chemoresistance, we performed an unbiased genome-wide RNAi screen in SLFN11-WT and -knockout (KO) cells. We found that inactivation of Ataxia Telangiectasia- and Rad3-related (ATR), CHK1, BRCA2, and RPA1 overcome chemoresistance to camptothecin (CPT) in SLFN11-KO cells. Accordingly, we validate that clinical inhibitors of ATR (M4344 and M6620) and CHK1 (SRA737) resensitize SLFN11-KO cells to topotecan, indotecan, etoposide, cisplatin, and talazoparib. We uncover that ATR inhibition significantly increases mitotic defects along with increased CDT1 phosphorylation, which destabilizes kinetochore-microtubule attachments in SLFN11-KO cells. We also reveal a chemoresistance mechanism by which CDT1 degradation is retarded, eventually inducing replication reactivation under DNA damage in SLFN11-KO cells. In contrast, in SLFN11-expressing cells, SLFN11 promotes the degradation of CDT1 in response to CPT by binding to DDB1 of CUL4CDT2 E3 ubiquitin ligase associated with replication forks. We show that the C terminus and ATPase domain of SLFN11 are required for DDB1 binding and CDT1 degradation. Furthermore, we identify a therapy-relevant ATPase mutant (E669K) of the SLFN11 gene in human TCGA and show that the mutant contributes to chemoresistance and retarded CDT1 degradation. Taken together, our study reveals new chemotherapeutic insights on how targeting the ATR pathway overcomes chemoresistance of SLFN11-deficient cancers. It also demonstrates that SLFN11 irreversibly arrests replication by degrading CDT1 through the DDB1-CUL4CDT2 ubiquitin ligase.
Keywords: ATR/CHK1 inhibitor; CDT1; CUL4; SLFN11.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
