

A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits
- PMID: 33536417
- PMCID: PMC7858636
- DOI: 10.1038/s41467-020-20885-8
A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits
Abstract
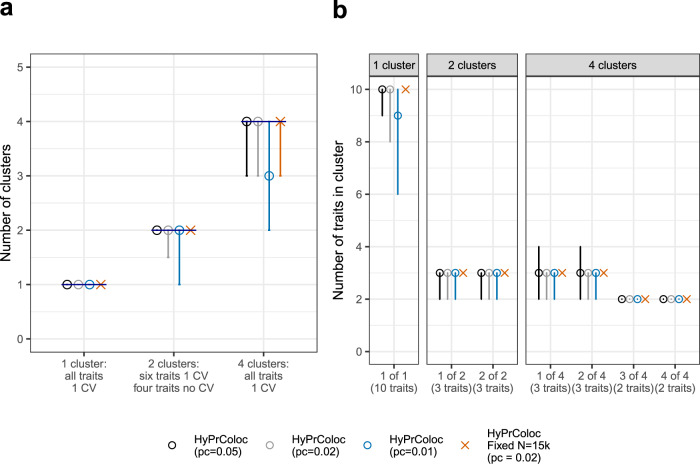
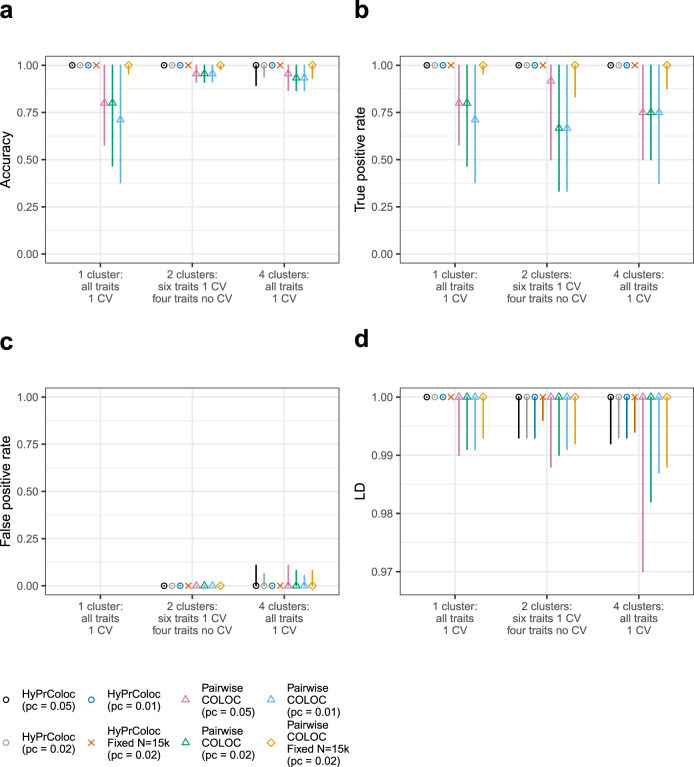
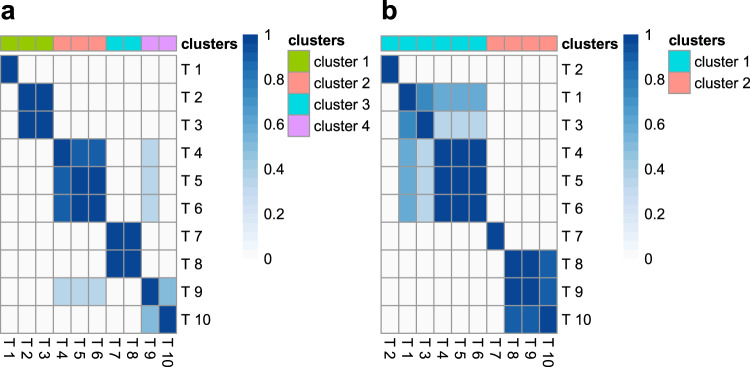
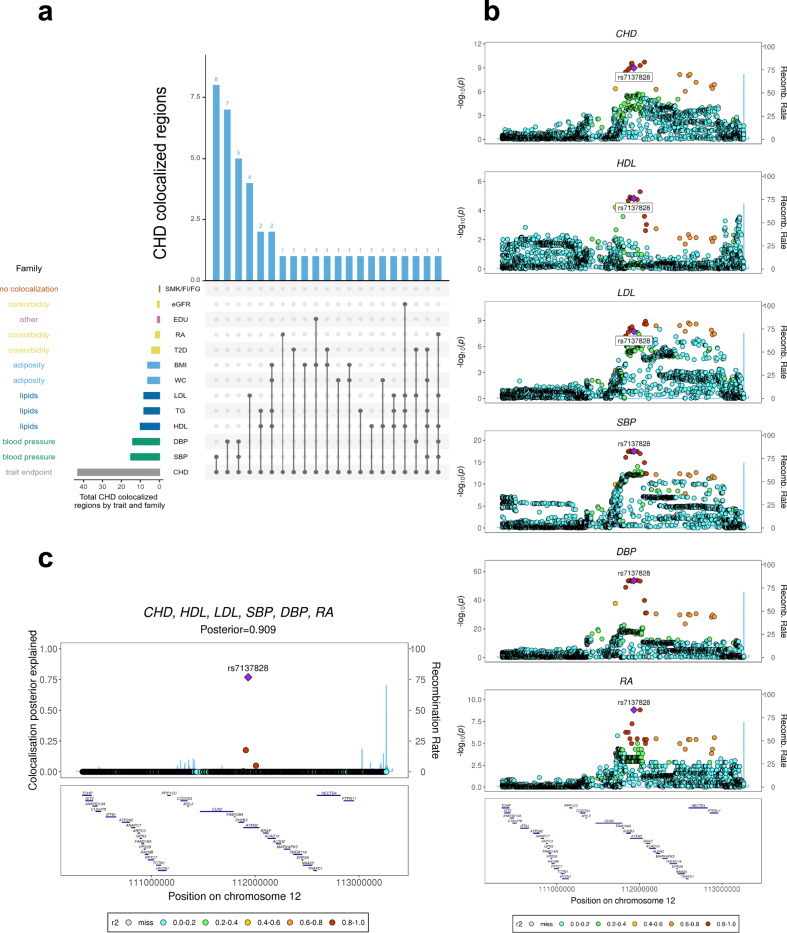
Genome-wide association studies (GWAS) have identified thousands of genomic regions affecting complex diseases. The next challenge is to elucidate the causal genes and mechanisms involved. One approach is to use statistical colocalization to assess shared genetic aetiology across multiple related traits (e.g. molecular traits, metabolic pathways and complex diseases) to identify causal pathways, prioritize causal variants and evaluate pleiotropy. We propose HyPrColoc (Hypothesis Prioritisation for multi-trait Colocalization), an efficient deterministic Bayesian algorithm using GWAS summary statistics that can detect colocalization across vast numbers of traits simultaneously (e.g. 100 traits can be jointly analysed in around 1 s). We perform a genome-wide multi-trait colocalization analysis of coronary heart disease (CHD) and fourteen related traits, identifying 43 regions in which CHD colocalized with ≥1 trait, including 5 previously unknown CHD loci. Across the 43 loci, we further integrate gene and protein expression quantitative trait loci to identify candidate causal genes.
Conflict of interest statement
J.M.M.H. became a full-time employee of Novo Nordisk Ltd while this manuscript was under review. All other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
