

Analysis of Fox genes in Schmidtea mediterranea reveals new families and a conserved role of Smed-foxO in controlling cell death
- PMID: 33536473
- PMCID: PMC7859237
- DOI: 10.1038/s41598-020-80627-0
Analysis of Fox genes in Schmidtea mediterranea reveals new families and a conserved role of Smed-foxO in controlling cell death
Abstract
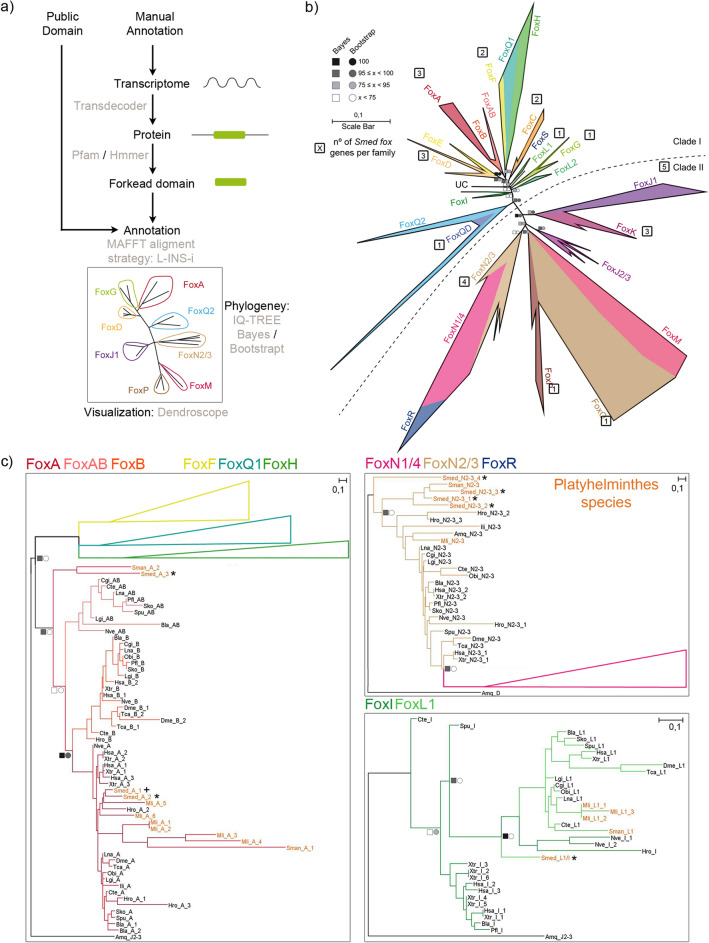
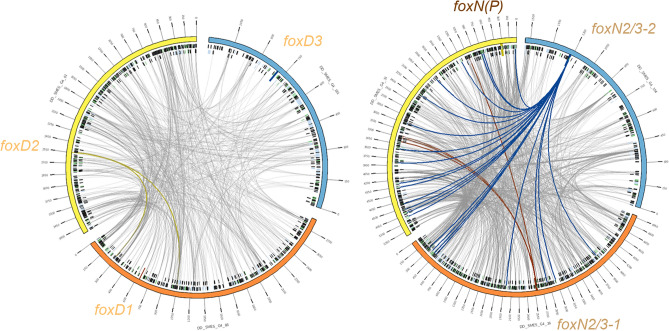
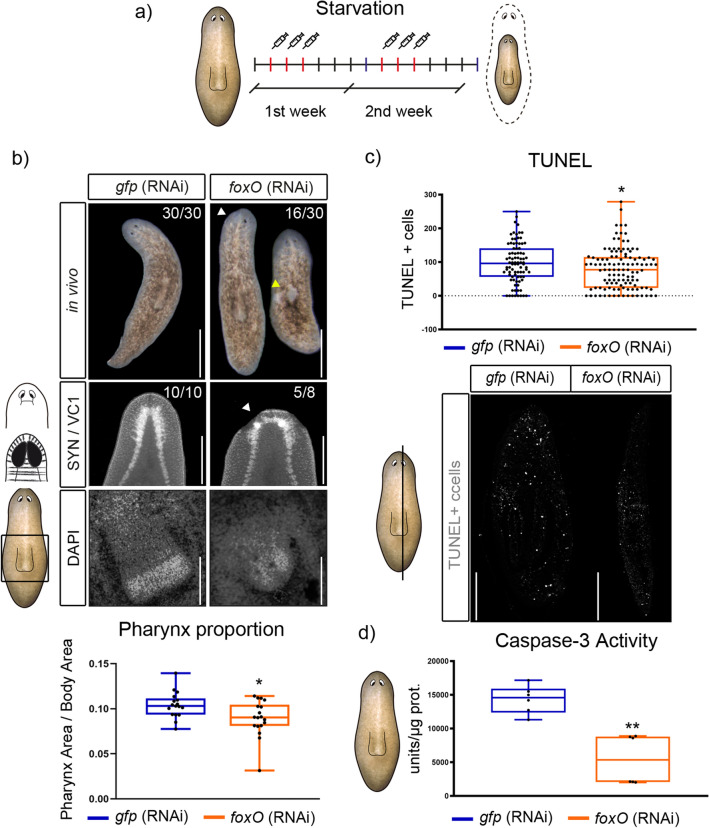
The forkhead box (Fox) genes encode transcription factors that control several key aspects of development. Present in the ancestor of all eukaryotes, Fox genes underwent several duplications followed by loss and diversification events that gave rise to the current 25 families. However, few Fox members have been identified from the Lophotrochozoa clade, and specifically from planarians, which are a unique model for understanding development, due to the striking plasticity of the adult. The aim of this study was to identify and perform evolutionary and functional studies of the Fox genes of lophotrochozoan species and, specifically, of the planarian Schmidtea mediterranea. Generating a pipeline for identifying Forkhead domains and using phylogenetics allowed us the phylogenetic reconstruction of Fox genes. We corrected the annotation for misannotated genes and uncovered a new family, the QD, present in all metazoans. According to the new phylogeny, the 27 Fox genes found in Schmidtea mediterranea were classified into 12 families. In Platyhelminthes, family losses were accompanied by extensive gene diversification and the appearance of specific families, the A(P) and N(P). Among the newly identified planarian Fox genes, we found a single copy of foxO, which shows an evolutionary conserved role in controlling cell death.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
