

RasGRP2 inhibits glyceraldehyde-derived toxic advanced glycation end-products from inducing permeability in vascular endothelial cells
- PMID: 33536515
- PMCID: PMC7859393
- DOI: 10.1038/s41598-021-82619-0
RasGRP2 inhibits glyceraldehyde-derived toxic advanced glycation end-products from inducing permeability in vascular endothelial cells
Abstract
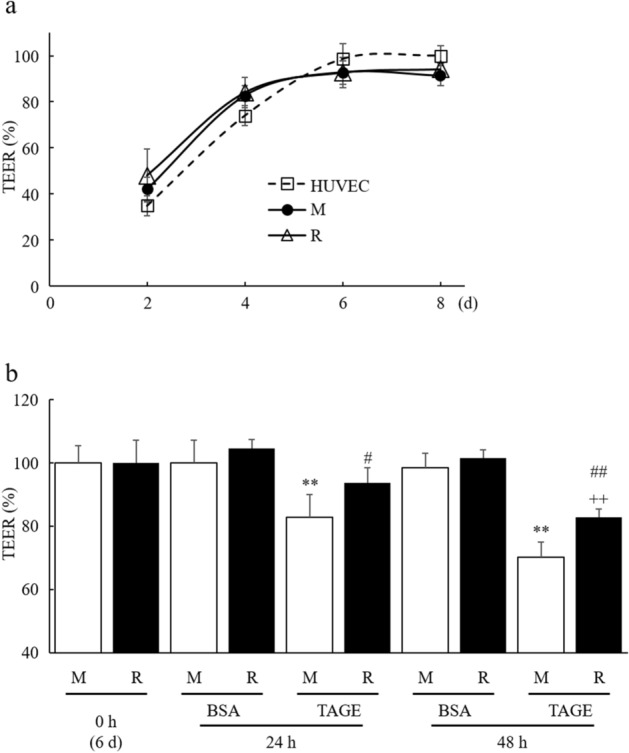
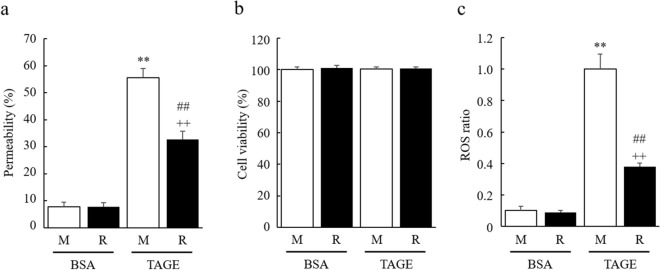
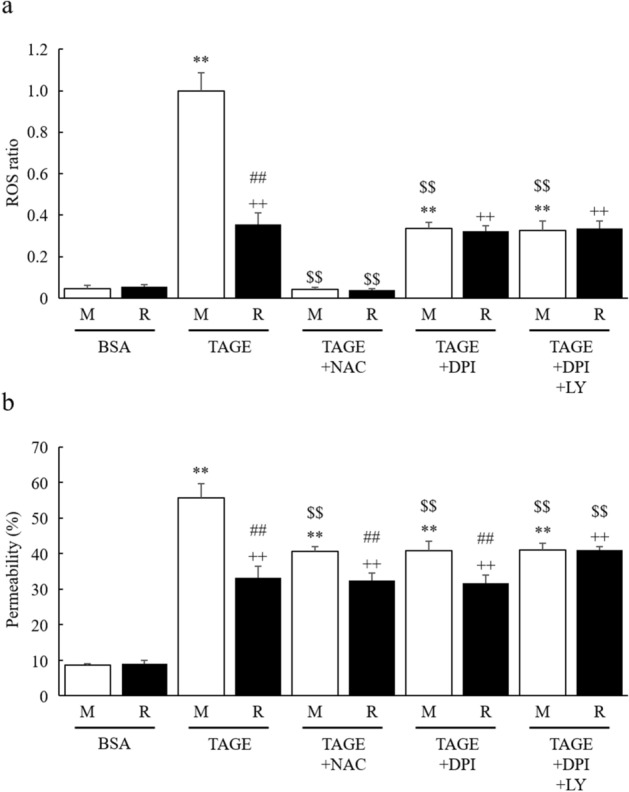
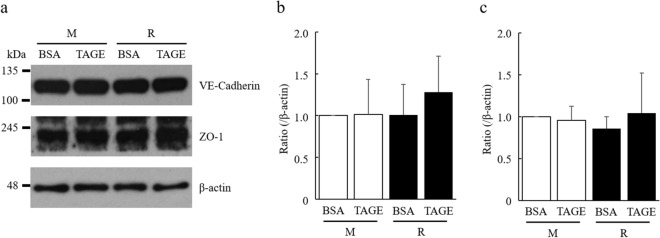
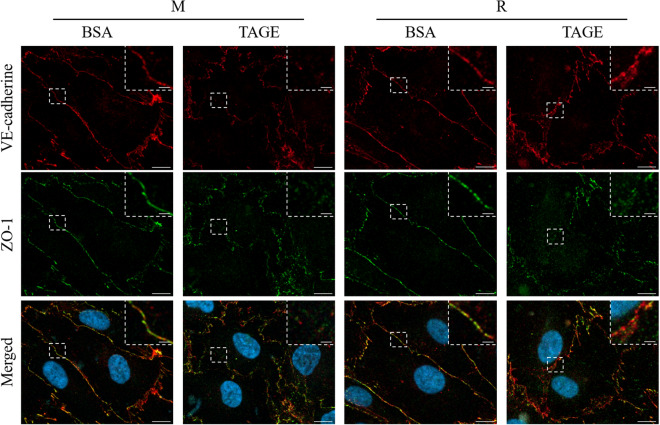
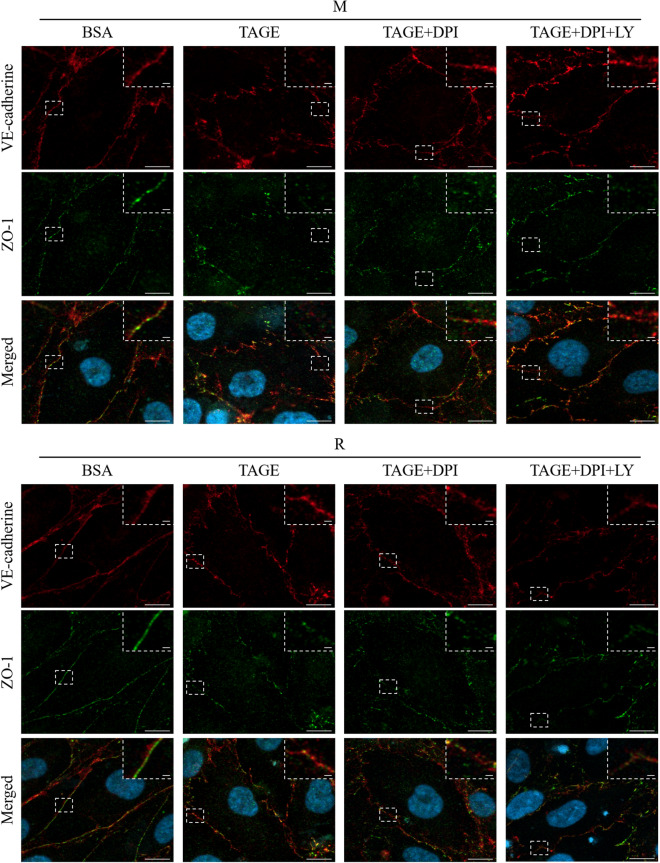
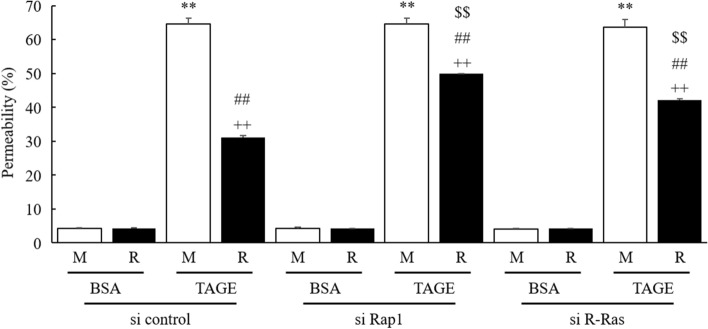
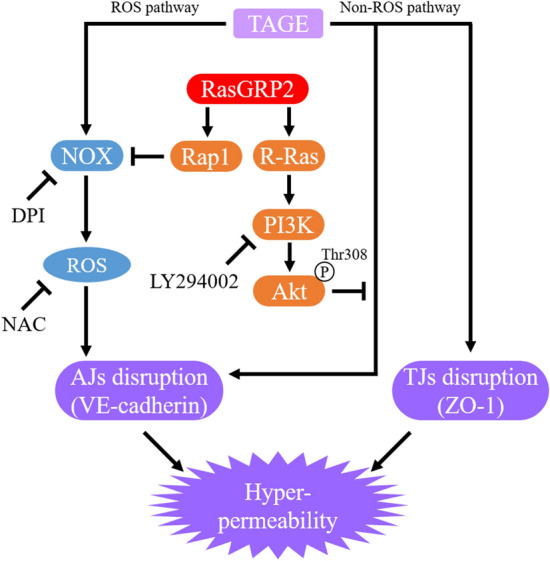
Advanced glycation end-products (AGEs) are formed by the non-enzymatic reaction of sugars and proteins. Among the AGEs, glyceraldehyde-derived toxic AGEs (TAGE) are associated with various diseases, including diabetic complications such as diabetic retinopathy (DR). The risk of developing DR is strongly associated with poor glycemic control, which causes AGE accumulation and increases AGE-induced vascular permeability. We previously reported that Ras guanyl nucleotide releasing protein 2 (RasGRP2), which activates small G proteins, may play an essential role in the cell response to toxicity when exposed to various factors. However, it is not known whether RasGRP2 prevents the adverse effects of TAGE in vascular endothelial cells. This study observed that TAGE enhanced vascular permeability by disrupting adherens junctions and tight junctions via complex signaling, such as ROS and non-ROS pathways. In particular, RasGRP2 protected adherens junction disruption, thereby suppressing vascular hyper-permeability. These results indicate that RasGRP2 is an essential protective factor of vascular permeability and may help develop novel therapeutic strategies for AGE-induced DR.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
