

IgA transcytosis and antigen recognition govern ovarian cancer immunity
- PMID: 33536615
- PMCID: PMC7969354
- DOI: 10.1038/s41586-020-03144-0
IgA transcytosis and antigen recognition govern ovarian cancer immunity
Abstract
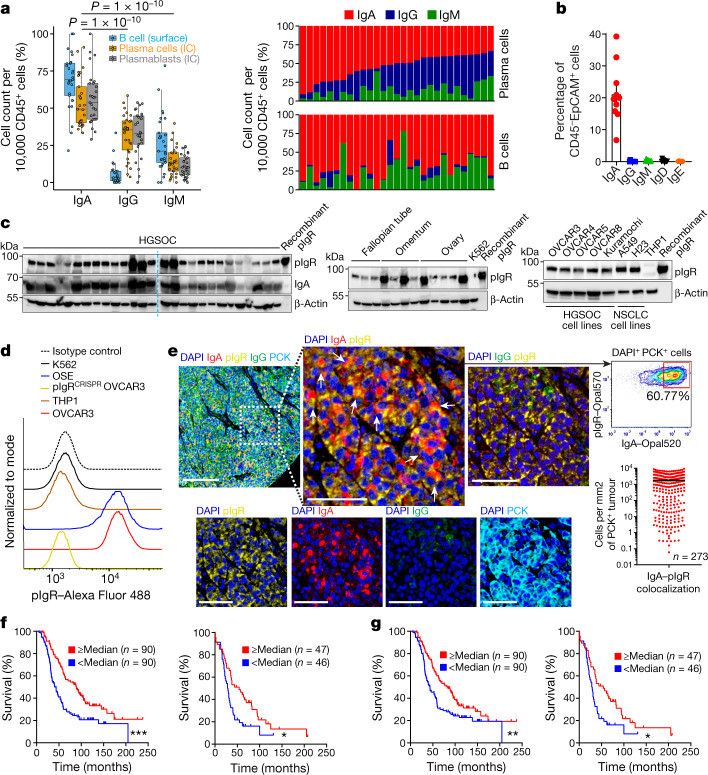
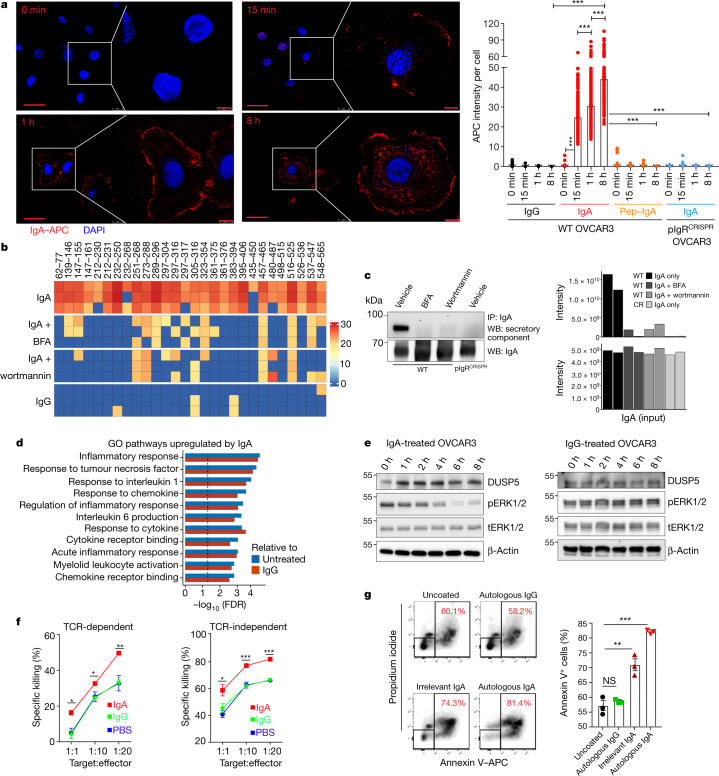
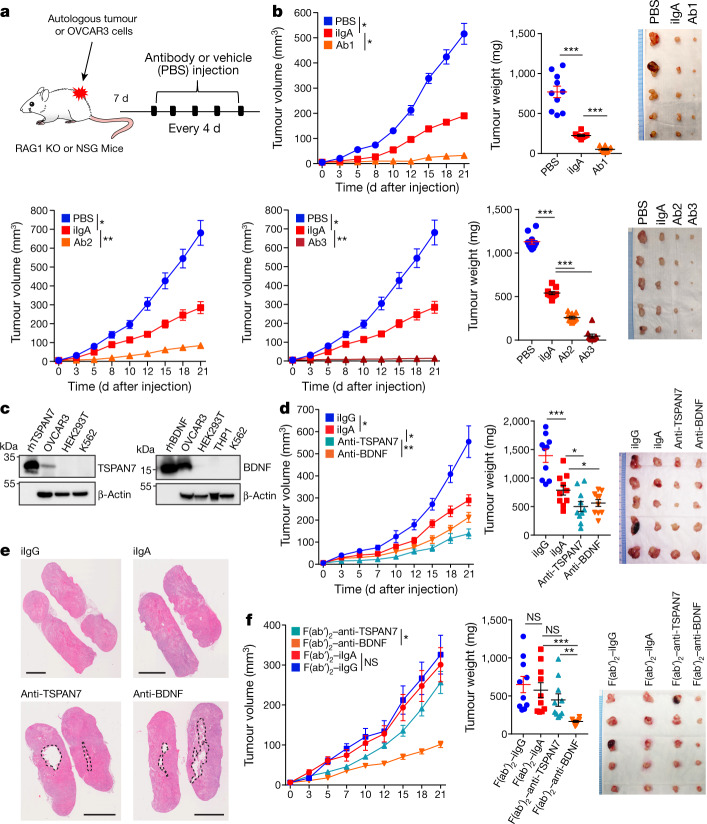
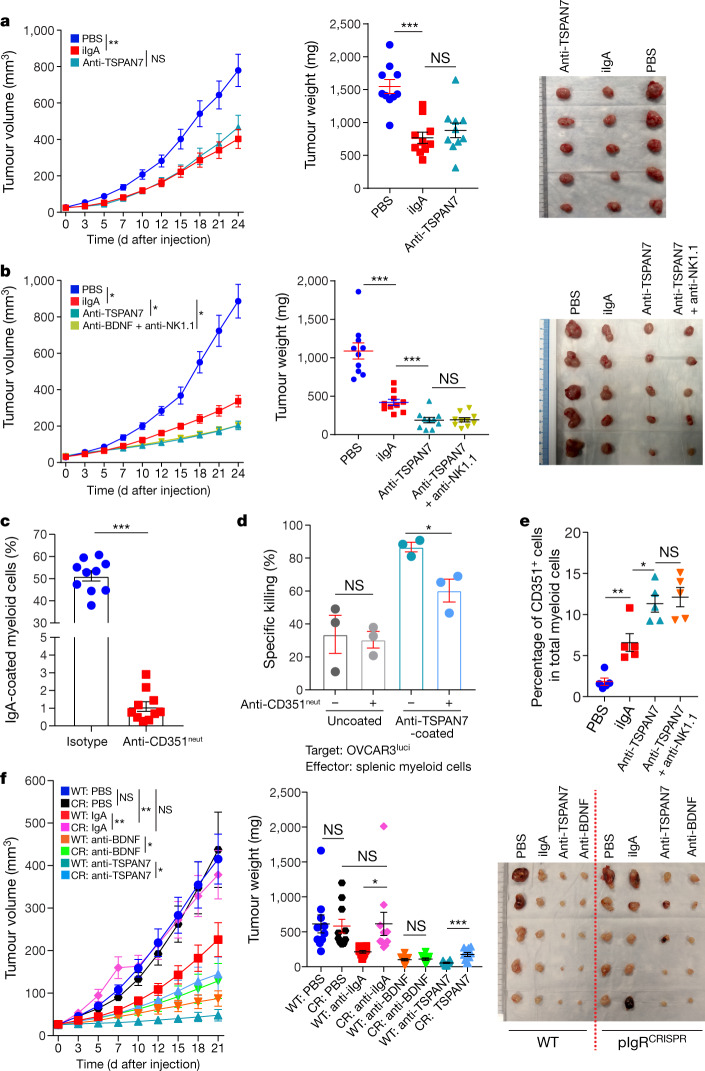
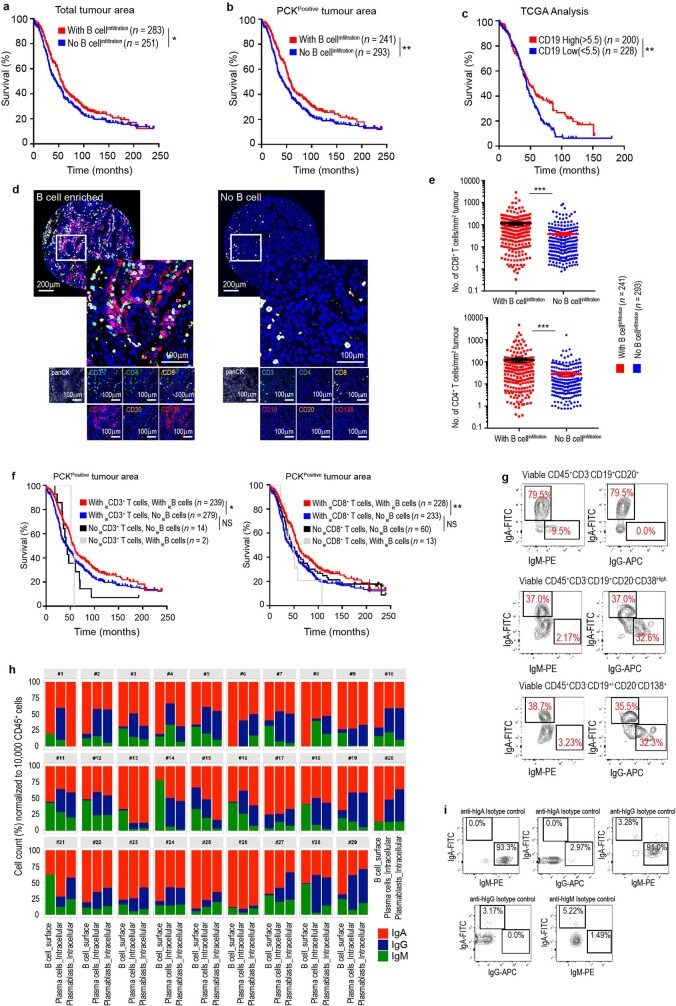
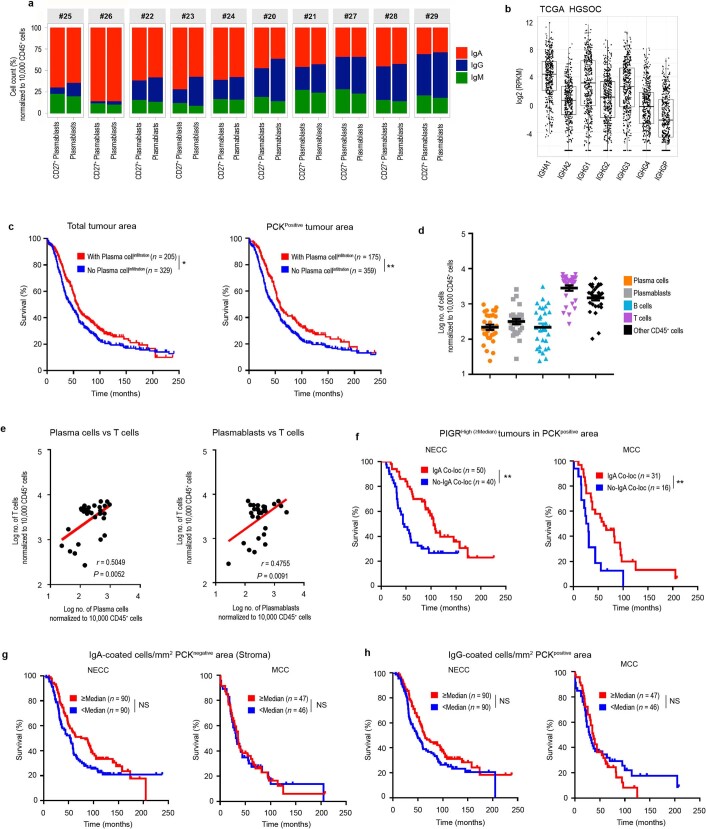
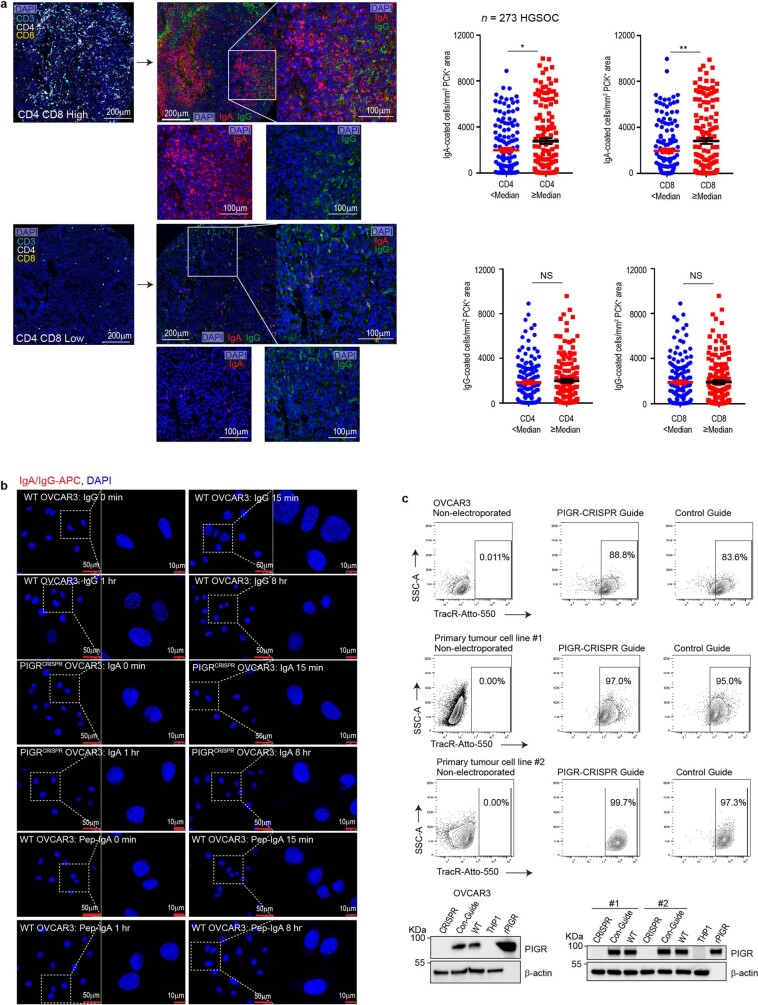
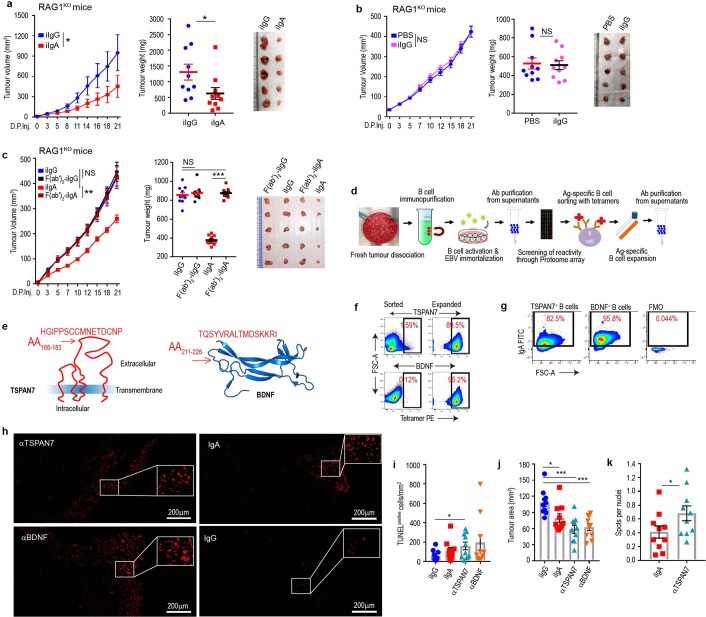
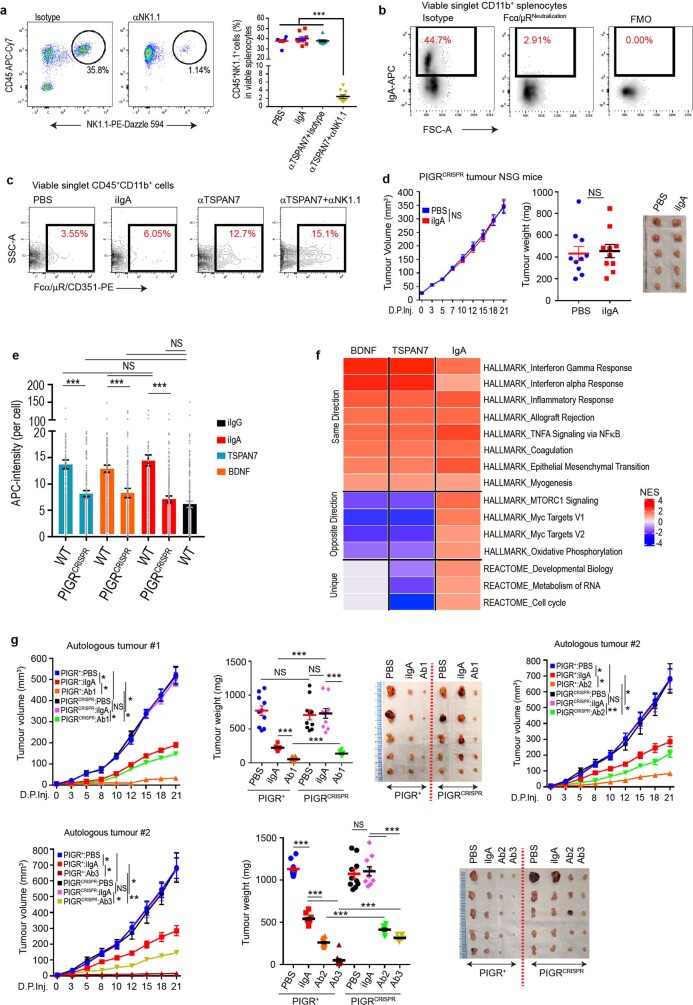
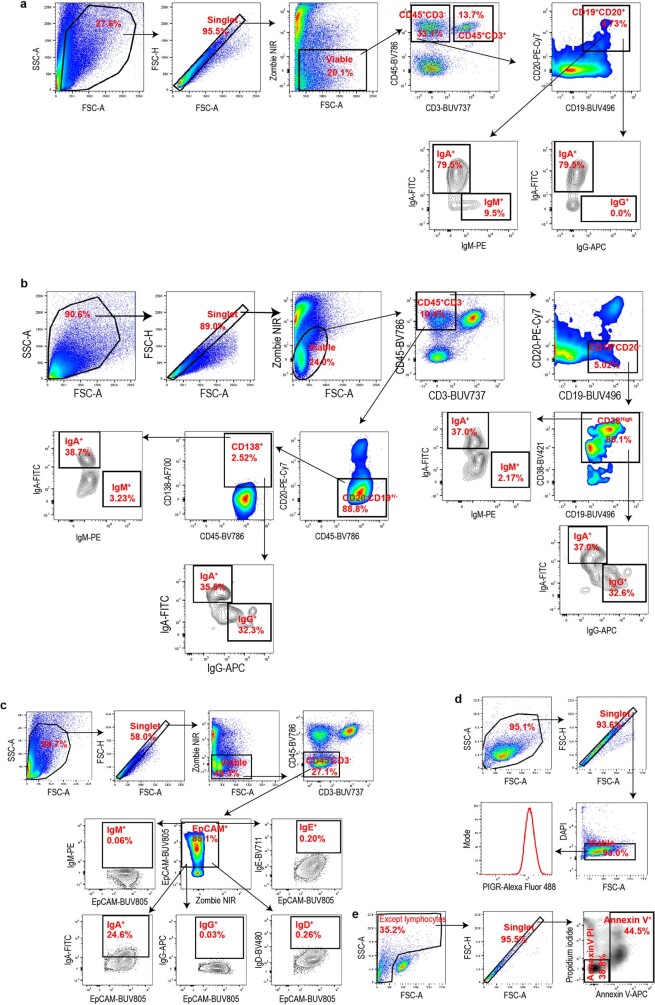
Most ovarian cancers are infiltrated by prognostically relevant activated T cells1-3, yet exhibit low response rates to immune checkpoint inhibitors4. Memory B cell and plasma cell infiltrates have previously been associated with better outcomes in ovarian cancer5,6, but the nature and functional relevance of these responses are controversial. Here, using 3 independent cohorts that in total comprise 534 patients with high-grade serous ovarian cancer, we show that robust, protective humoral responses are dominated by the production of polyclonal IgA, which binds to polymeric IgA receptors that are universally expressed on ovarian cancer cells. Notably, tumour B-cell-derived IgA redirects myeloid cells against extracellular oncogenic drivers, which causes tumour cell death. In addition, IgA transcytosis through malignant epithelial cells elicits transcriptional changes that antagonize the RAS pathway and sensitize tumour cells to cytolytic killing by T cells, which also contributes to hindering malignant progression. Thus, tumour-antigen-specific and -antigen-independent IgA responses antagonize the growth of ovarian cancer by governing coordinated tumour cell, T cell and B cell responses. These findings provide a platform for identifying targets that are spontaneously recognized by intratumoural B-cell-derived antibodies, and suggest that immunotherapies that augment B cell responses may be more effective than approaches that focus on T cells, particularly for malignancies that are resistant to checkpoint inhibitors.
Conflict of interest statement
J.R.C.-G. has stock options and sponsored research, and receives consulting fees from Compass Therapeutics and Anixa Biosciences. He also receives consulting fees from Leidos. R.M.W. reports grants and personal fees from Merck, personal fees from Tesaro/GSK, personal fees from Genentech, personal fees from Legend Biotech, personal fees from AbbVie, personal fees from AstraZeneca, grants and stock from Ovation Diagnostics, personal fees from Clovis Oncology and personal fees from Regeneron, outside the submitted work. Otherwise, the authors do not have any conflicts of interest.
Figures

Comment in
-
IgA strikes twice in ovarian cancer.Nat Rev Cancer. 2021 Apr;21(4):215. doi: 10.1038/s41568-021-00342-4. Nat Rev Cancer. 2021. PMID: 33627799 No abstract available.
-
IgA transcytosis: A new weapon in the immune response to cancer?Cancer Cell. 2021 May 10;39(5):607-609. doi: 10.1016/j.ccell.2021.04.007. Cancer Cell. 2021. PMID: 33974859
References
MeSH terms
Substances
Grants and funding
- R01 CA233512/CA/NCI NIH HHS/United States
- P01 CA087969/CA/NCI NIH HHS/United States
- UM1 CA176726/CA/NCI NIH HHS/United States
- P30 CA076292/CA/NCI NIH HHS/United States
- UM1 CA186107/CA/NCI NIH HHS/United States
- R01 CA157664/CA/NCI NIH HHS/United States
- R01 CA262121/CA/NCI NIH HHS/United States
- R01 CA178687/CA/NCI NIH HHS/United States
- U01 CA232758/CA/NCI NIH HHS/United States
- U01 CA176726/CA/NCI NIH HHS/United States
- T32 CA009140/CA/NCI NIH HHS/United States
- P01 CA250984/CA/NCI NIH HHS/United States
- R35 CA197605/CA/NCI NIH HHS/United States
- R01 CA211913/CA/NCI NIH HHS/United States
- R01 CA124515/CA/NCI NIH HHS/United States
- R01 CA054419/CA/NCI NIH HHS/United States
- R01 CA184185/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
