

A gene-environment-induced epigenetic program initiates tumorigenesis
- PMID: 33536616
- PMCID: PMC8482641
- DOI: 10.1038/s41586-020-03147-x
A gene-environment-induced epigenetic program initiates tumorigenesis
Abstract
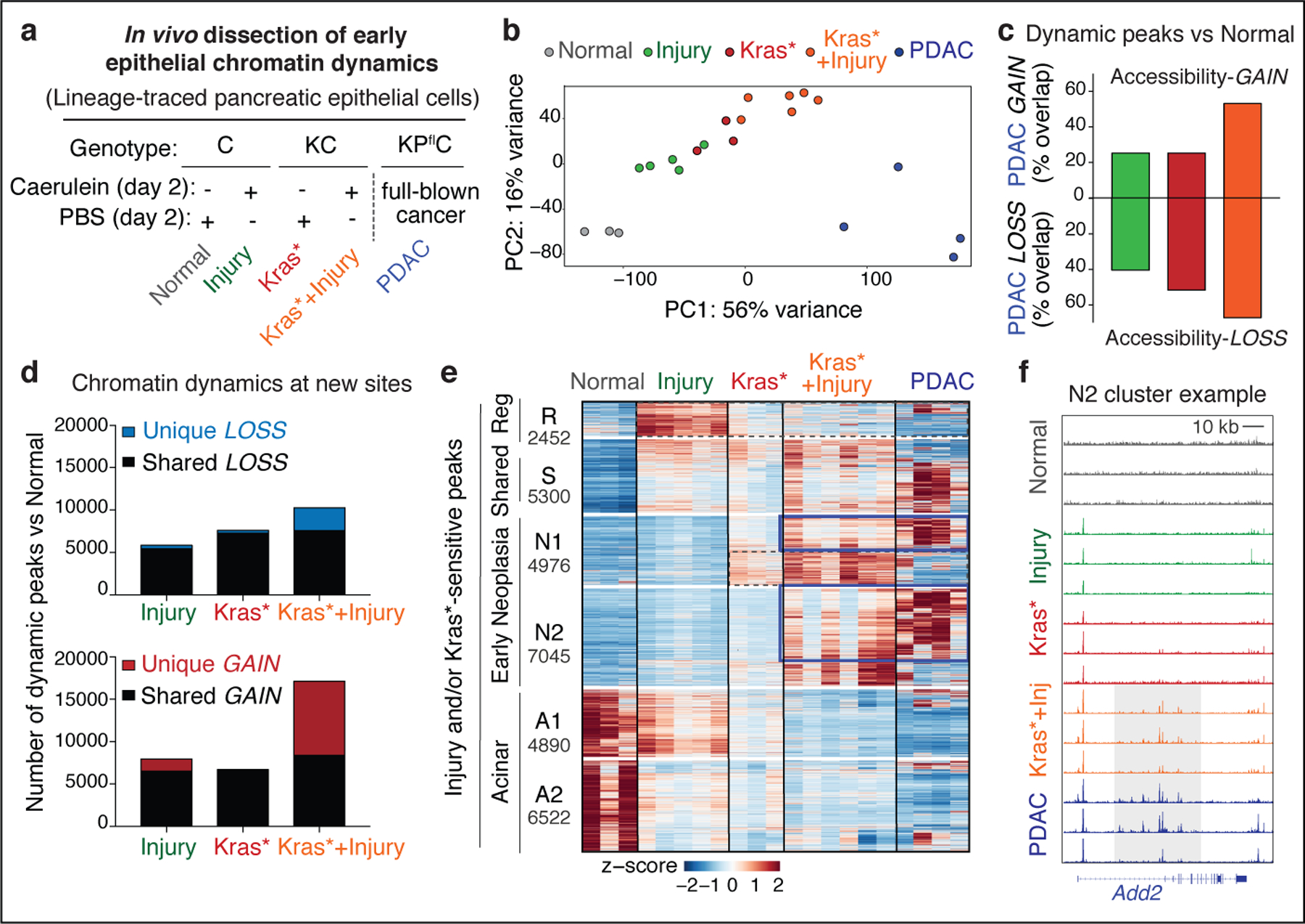
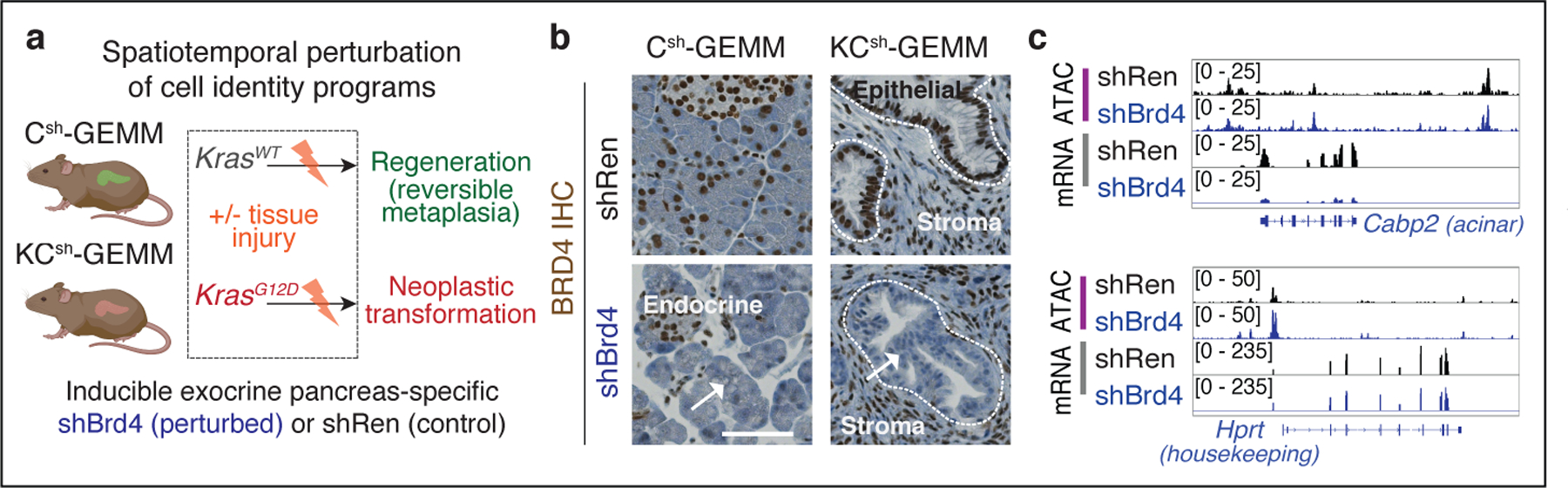
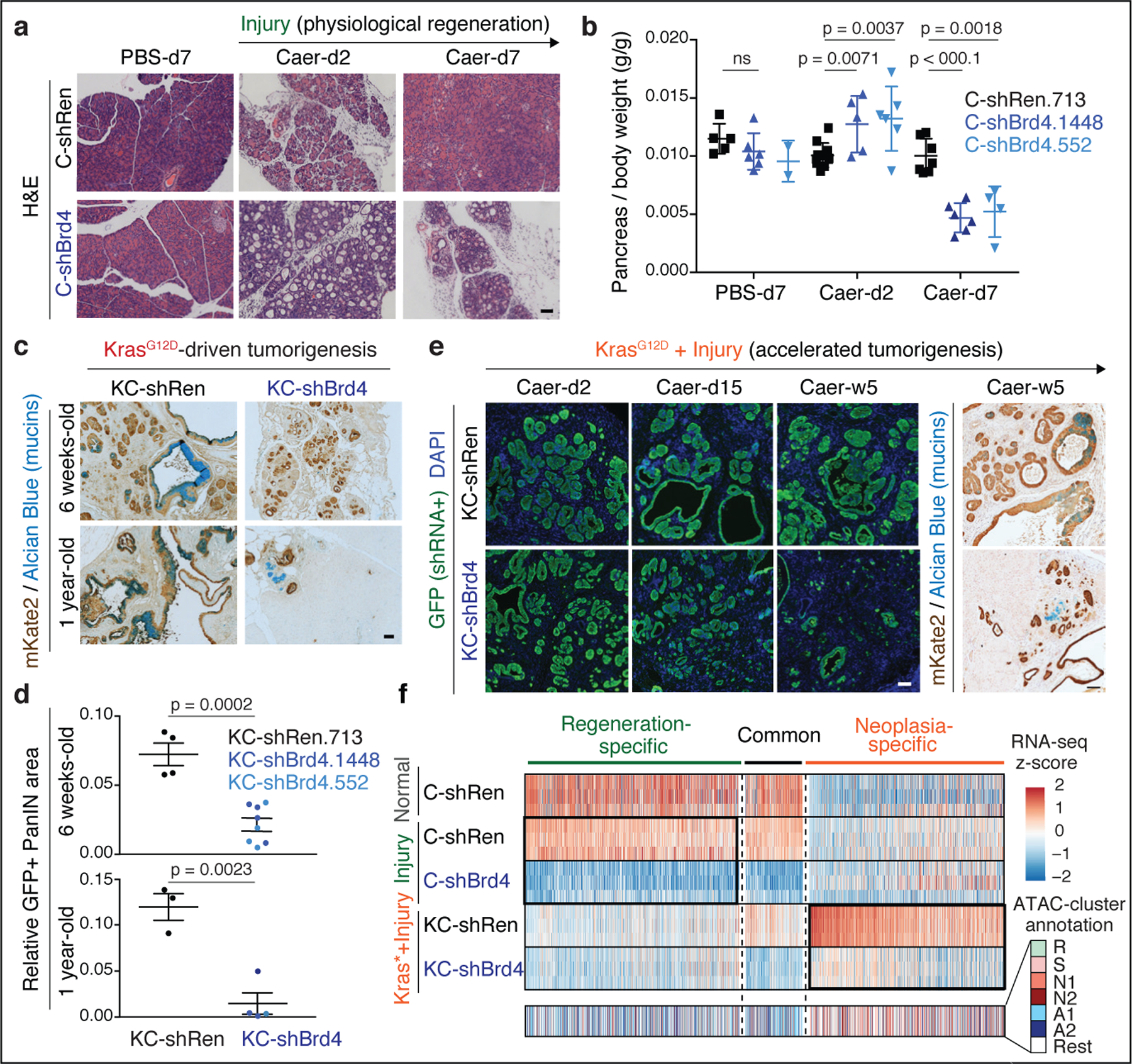
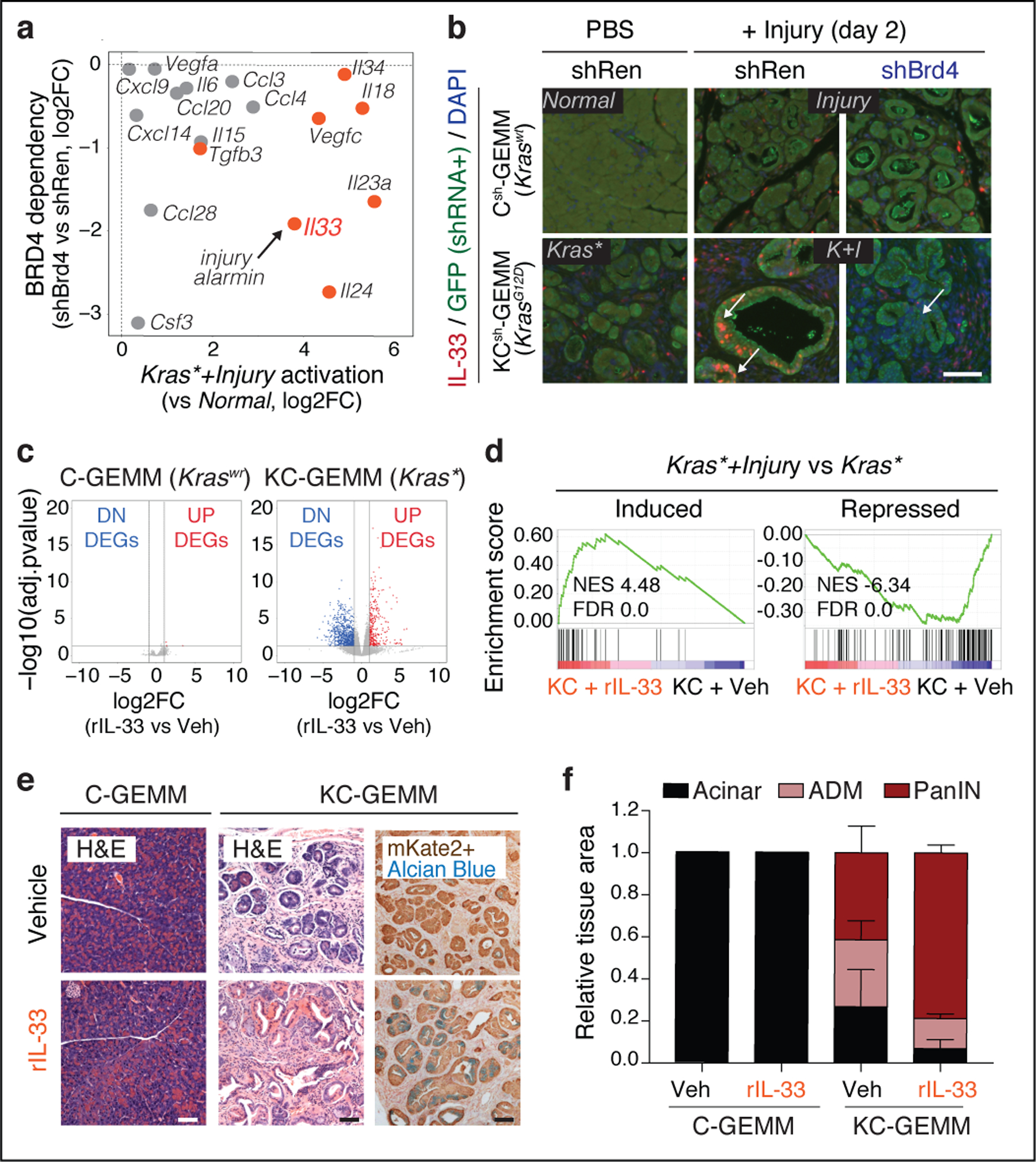
Tissue damage increases the risk of cancer through poorly understood mechanisms1. In mouse models of pancreatic cancer, pancreatitis associated with tissue injury collaborates with activating mutations in the Kras oncogene to markedly accelerate the formation of early neoplastic lesions and, ultimately, adenocarcinoma2,3. Here, by integrating genomics, single-cell chromatin assays and spatiotemporally controlled functional perturbations in autochthonous mouse models, we show that the combination of Kras mutation and tissue damage promotes a unique chromatin state in the pancreatic epithelium that distinguishes neoplastic transformation from normal regeneration and is selected for throughout malignant evolution. This cancer-associated epigenetic state emerges within 48 hours of pancreatic injury, and involves an 'acinar-to-neoplasia' chromatin switch that contributes to the early dysregulation of genes that define human pancreatic cancer. Among the factors that are most rapidly activated after tissue damage in the pre-malignant pancreatic epithelium is the alarmin cytokine interleukin 33, which recapitulates the effects of injury in cooperating with mutant Kras to unleash the epigenetic remodelling program of early neoplasia and neoplastic transformation. Collectively, our study demonstrates how gene-environment interactions can rapidly produce gene-regulatory programs that dictate early neoplastic commitment, and provides a molecular framework for understanding the interplay between genetic and environmental cues in the initiation of cancer.
Conflict of interest statement
Competing interest
A patent application (PTC/US2019/041670, internationally filing date 12 July 2019) has been submitted based in part on results presented in this manuscript covering methods for preventing or treating KRAS mutant pancreas cancer with inhibitors of Type 2 cytokine signaling. D.A.C and S.W.L are listed as the inventors. S.W.L. is a founder and scientific advisory board member of Blueprint Medicines, Mirimus Inc., and ORIC pharmaceuticals, and Faeth Therapeutics, and on the scientific advisory board of Constellation Pharmaceuticals and PMV Pharmaceuticals. S.D.L. is on the scientific advisory board of Nybo Therapeutics and Episteme Prognostics.
Figures
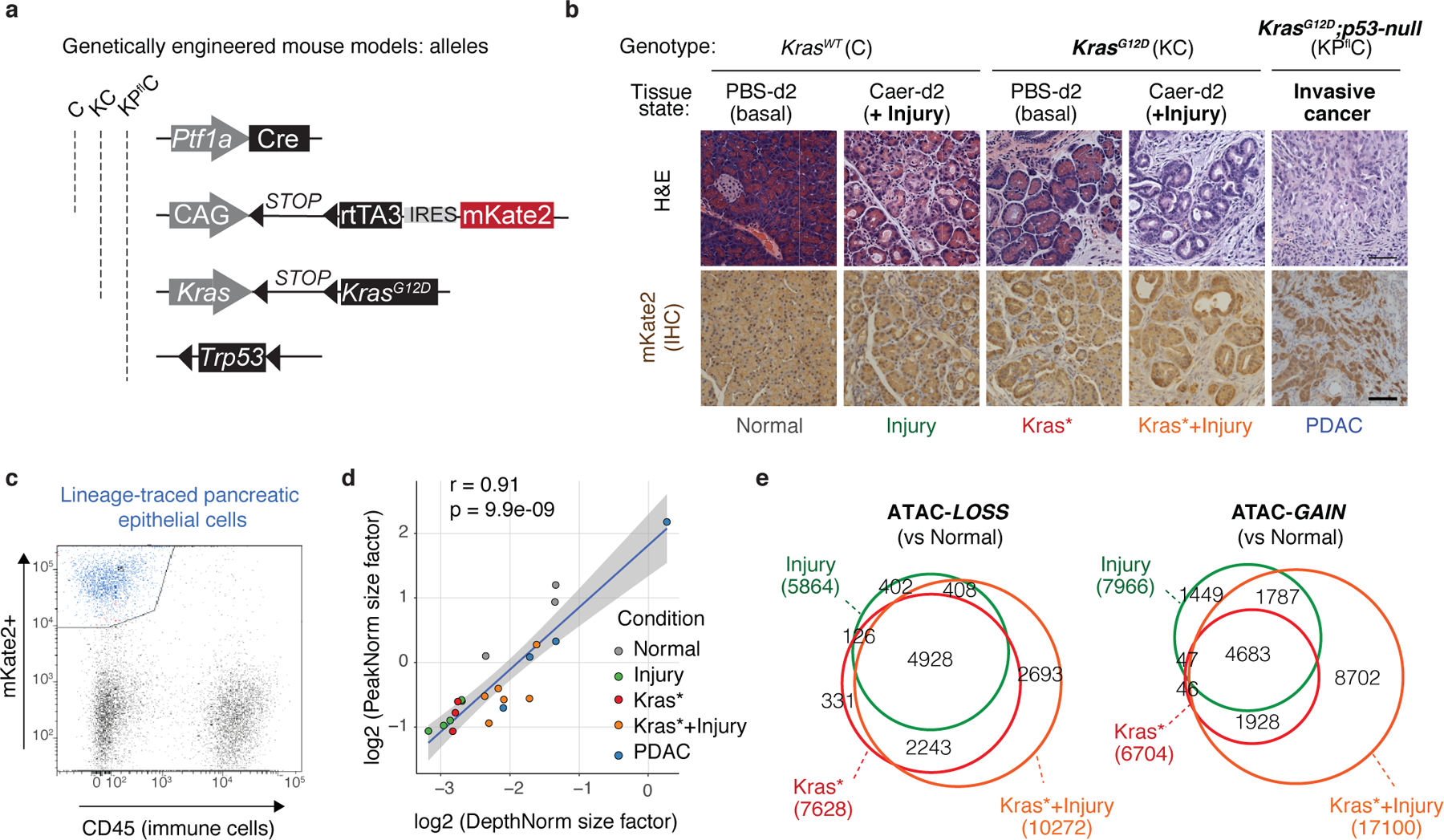

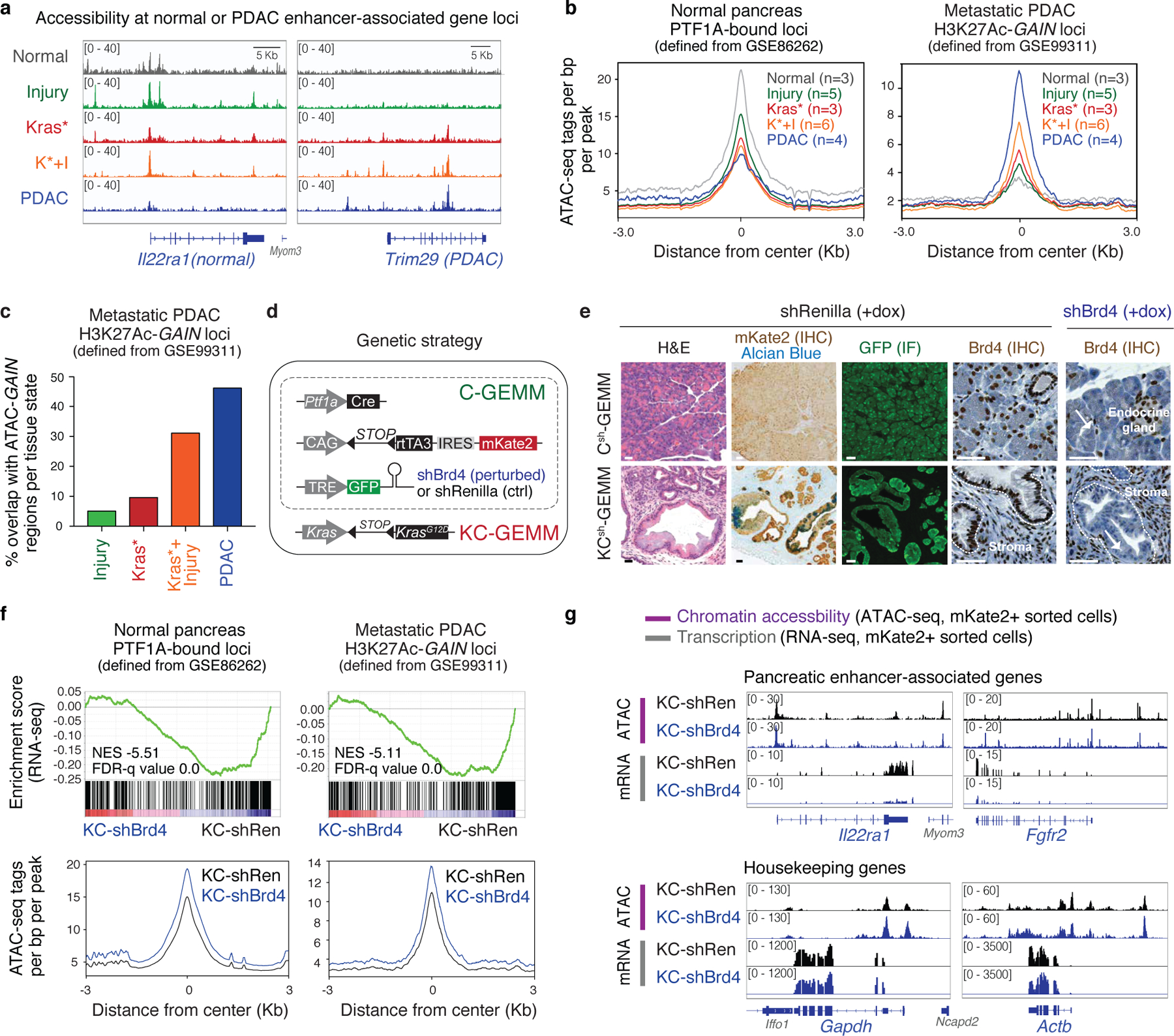
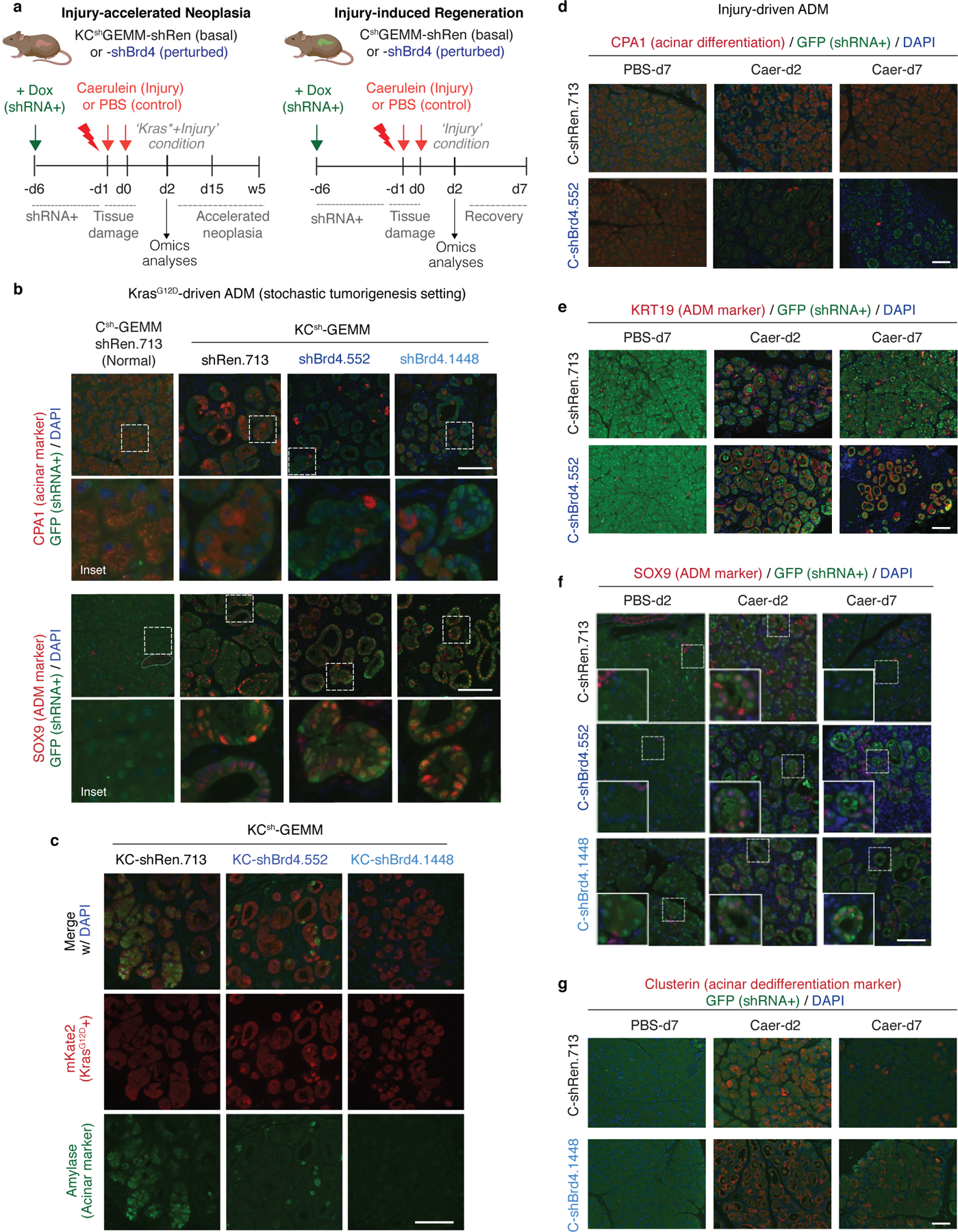
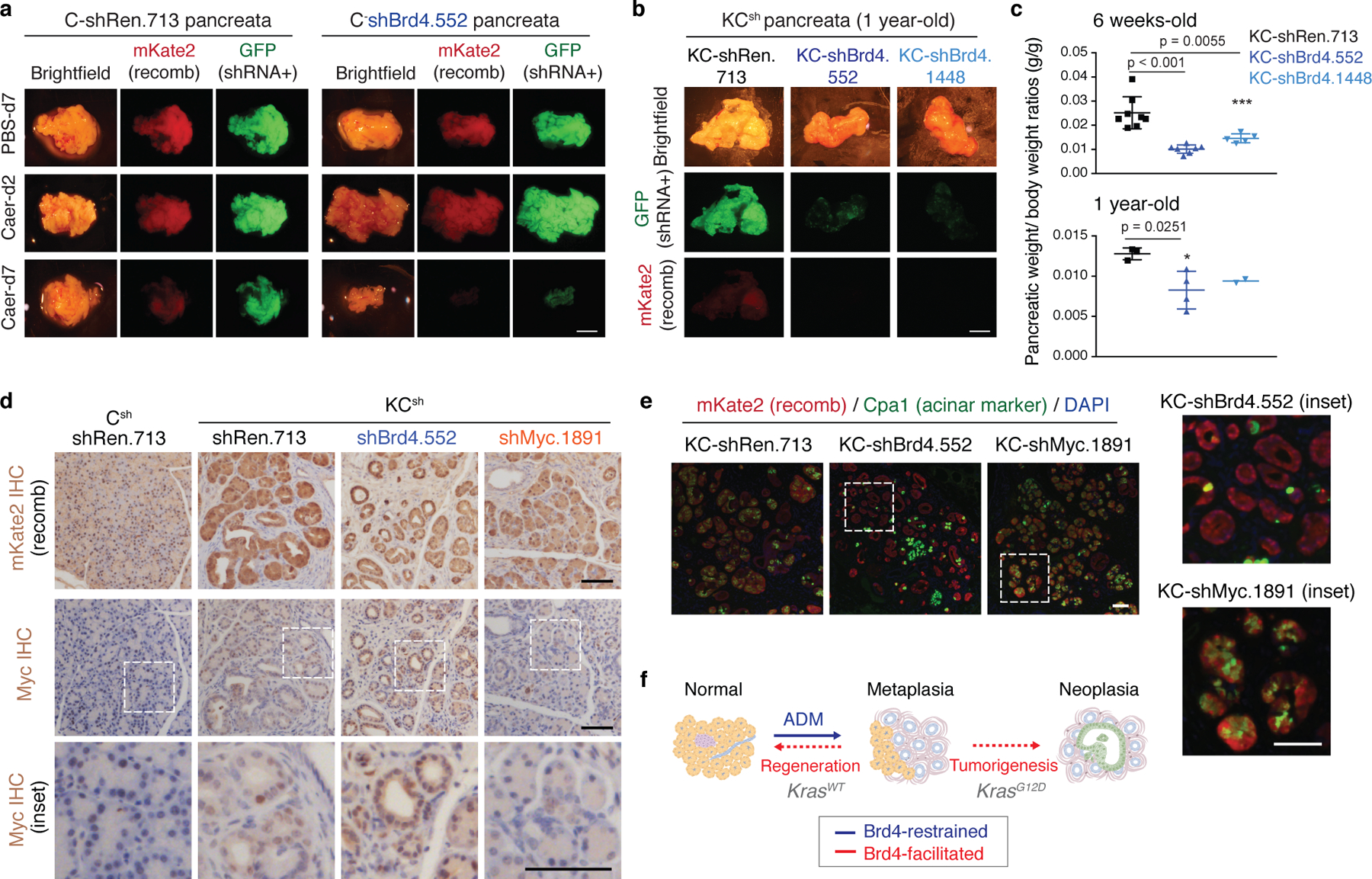
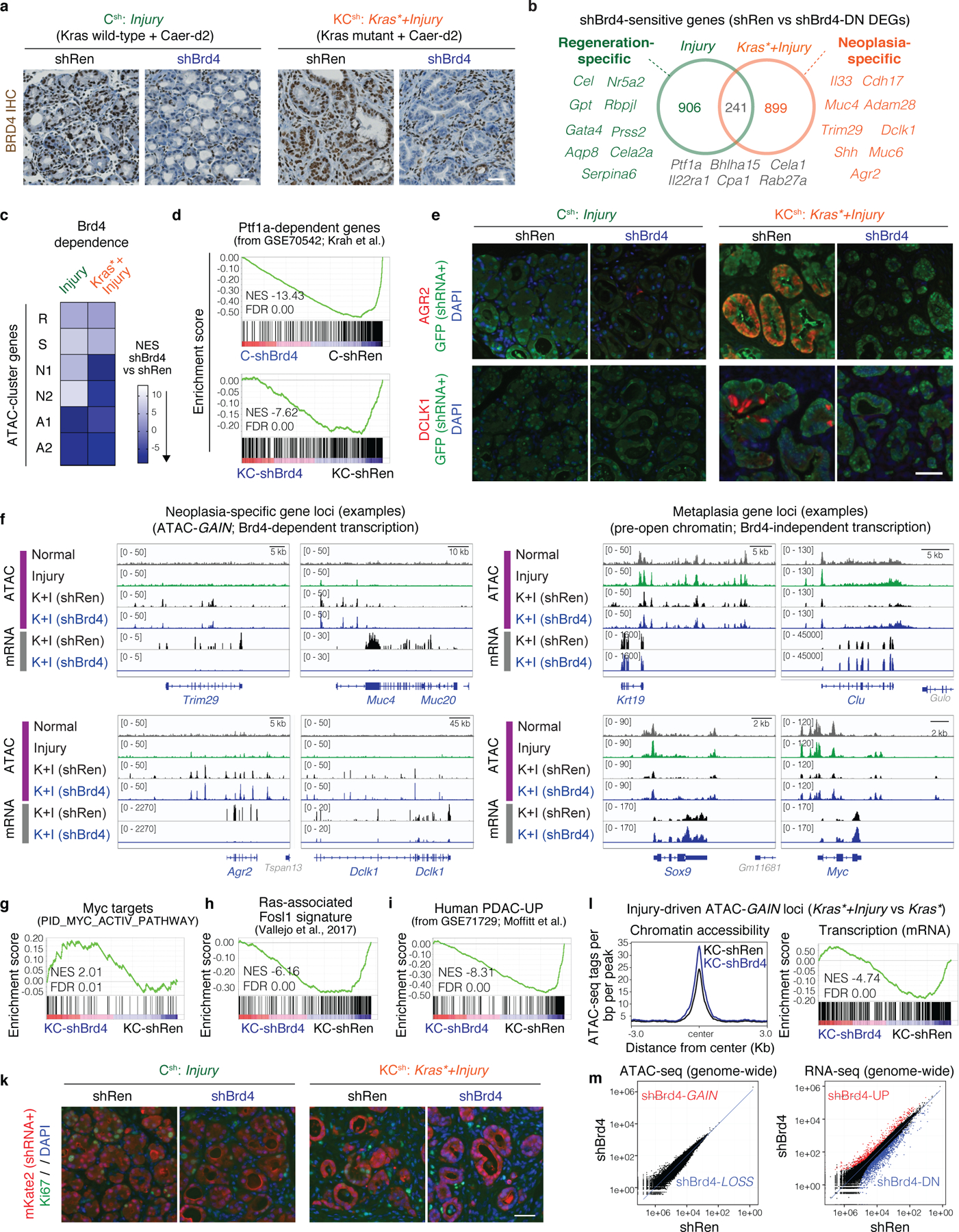
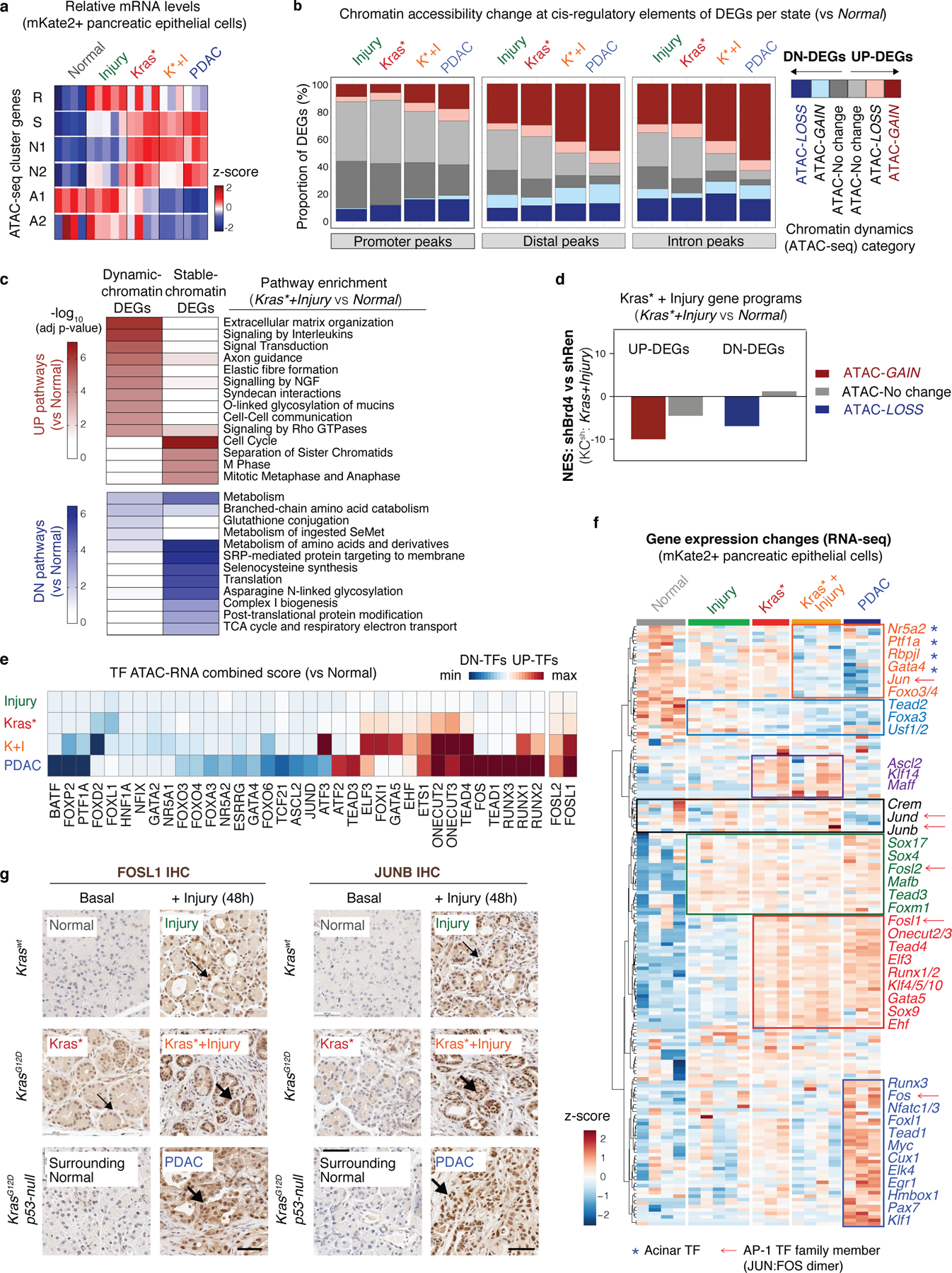
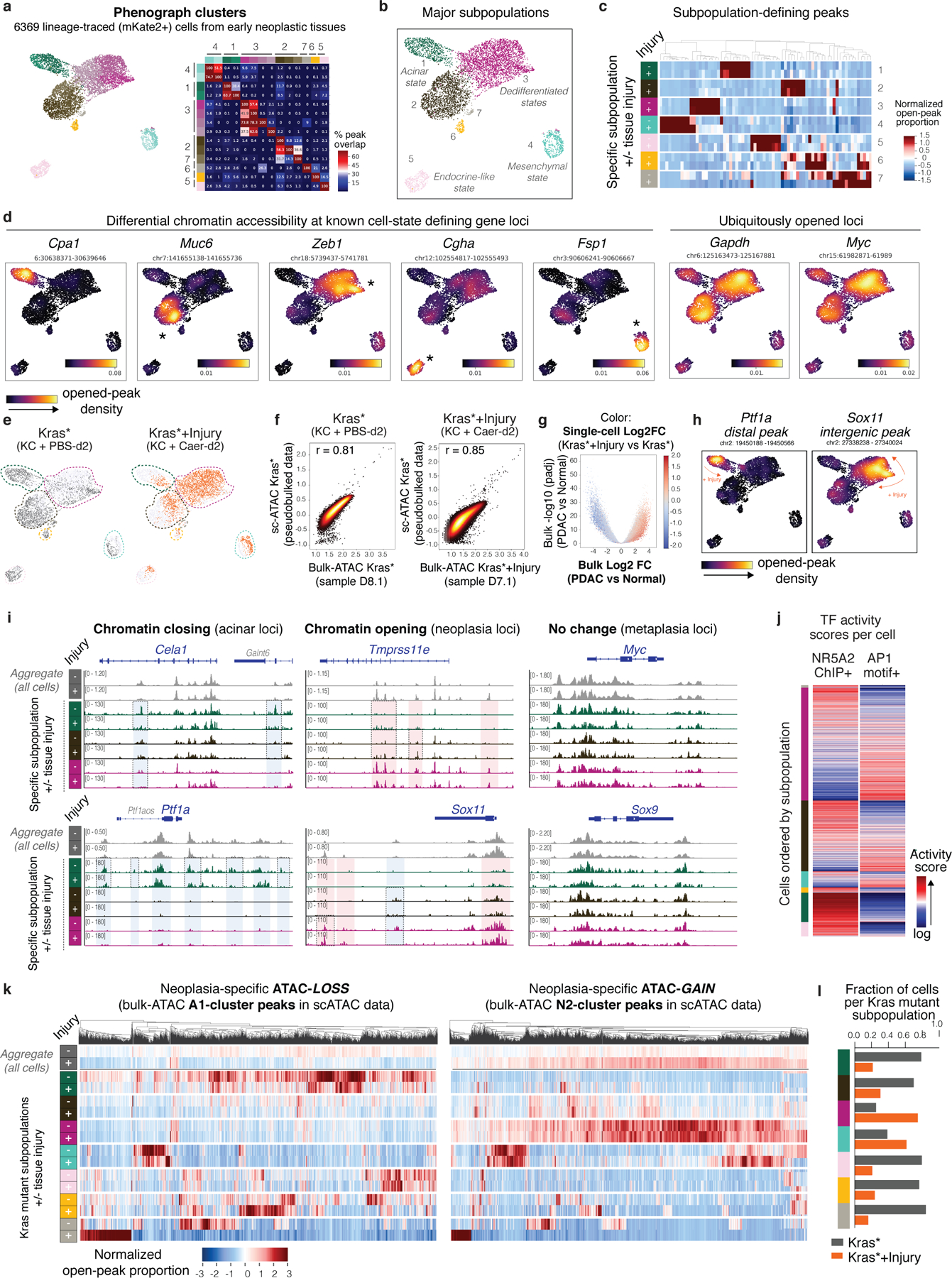
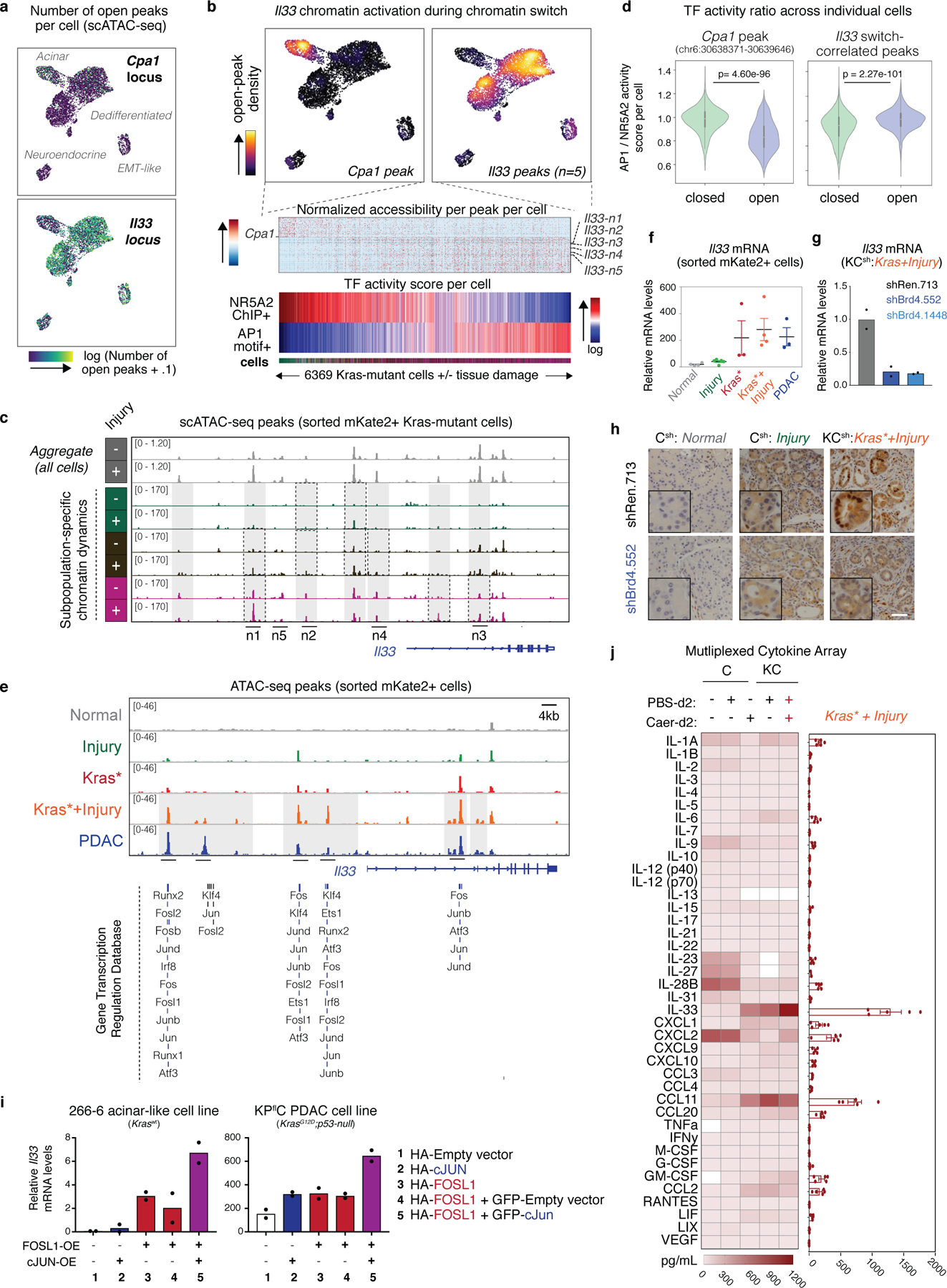


References
Main References
-
- Guerra C et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302, (2007). - PubMed
-
- Hingorani SR et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450 (2003). - PubMed
Methods References
Publication types
MeSH terms
Substances
Grants and funding
- T32 CA160001/CA/NCI NIH HHS/United States
- 5T32CA160001-08/NH/NIH HHS/United States
- F31 CA246901/CA/NCI NIH HHS/United States
- F32 CA177072/CA/NCI NIH HHS/United States
- P30 CA023108/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R00 CA230195/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- K99 CA230195/CA/NCI NIH HHS/United States
- K99 CA237736/CA/NCI NIH HHS/United States
- U54 CA209975/CA/NCI NIH HHS/United States
- R01 CA204228/CA/NCI NIH HHS/United States
- P01 CA013106/CA/NCI NIH HHS/United States
- F99 CA245797/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
