

Regulatory genomic circuitry of human disease loci by integrative epigenomics
- PMID: 33536621
- PMCID: PMC7875769
- DOI: 10.1038/s41586-020-03145-z
Regulatory genomic circuitry of human disease loci by integrative epigenomics
Erratum in
-
Author Correction: Regulatory genomic circuitry of human disease loci by integrative epigenomics.Nature. 2025 Jul;643(8071):E11. doi: 10.1038/s41586-025-09134-4. Nature. 2025. PMID: 40542162 Free PMC article. No abstract available.
Abstract
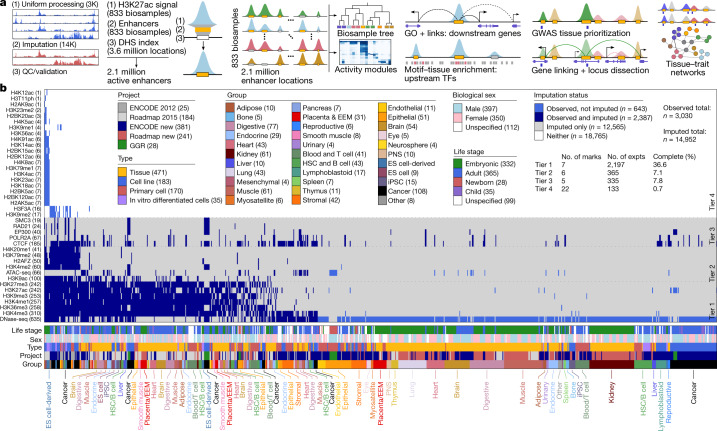
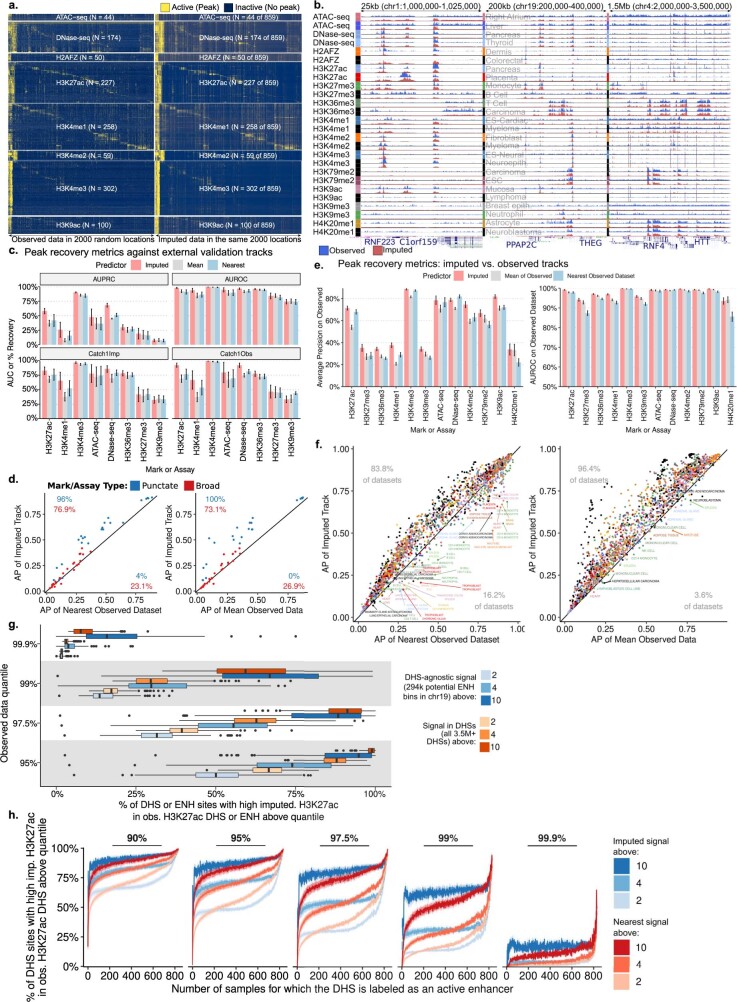
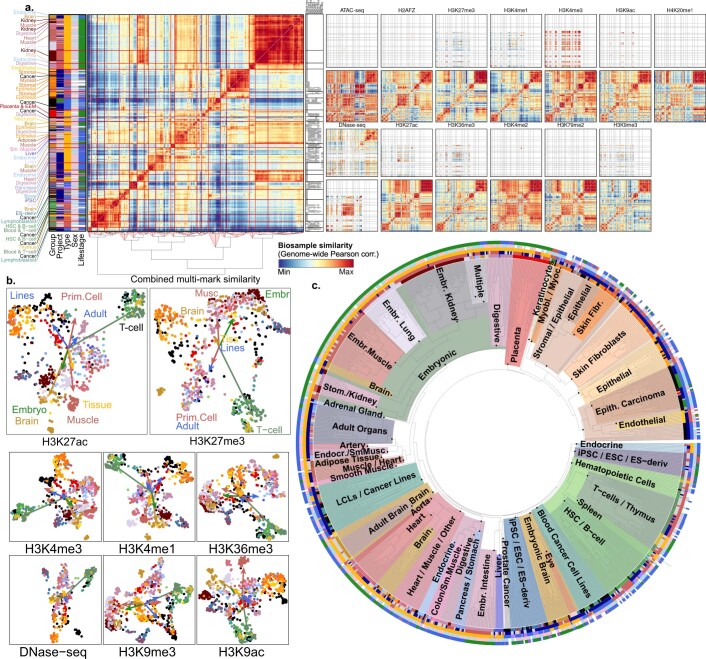
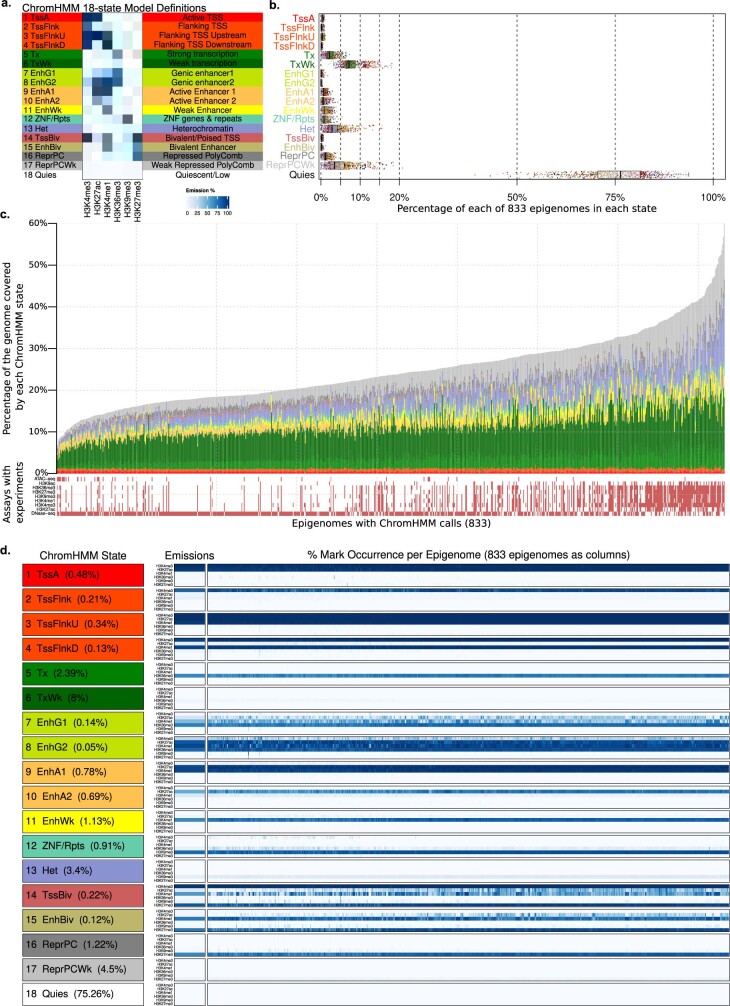
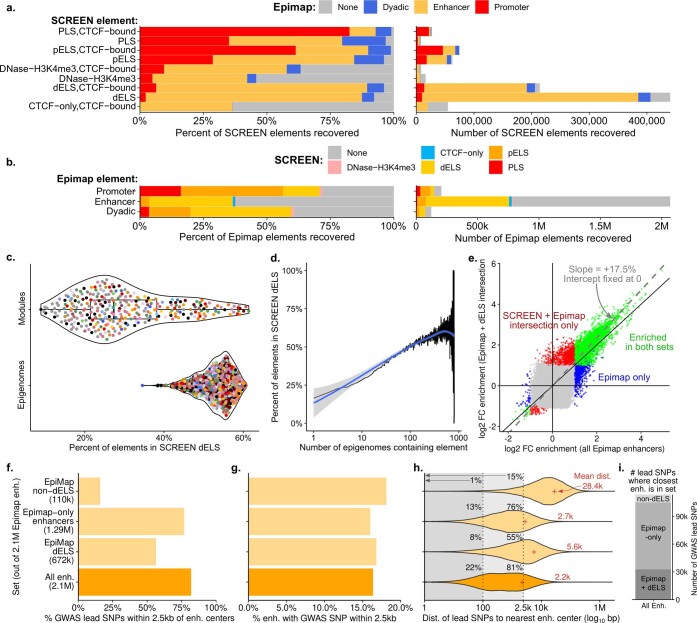
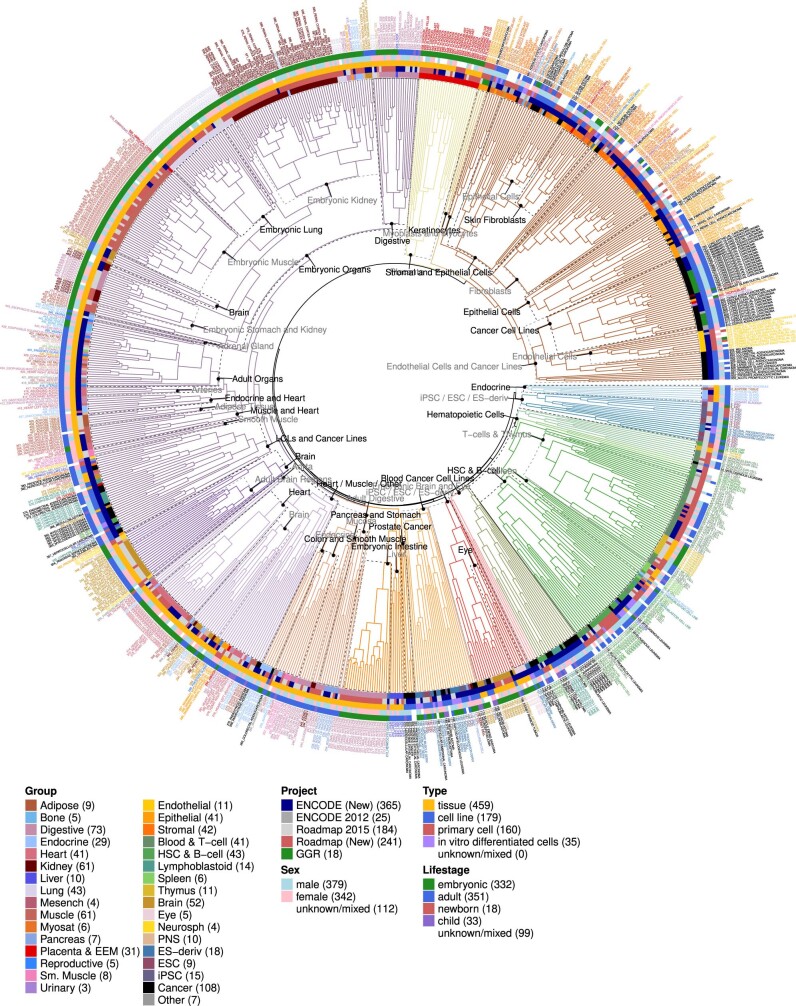
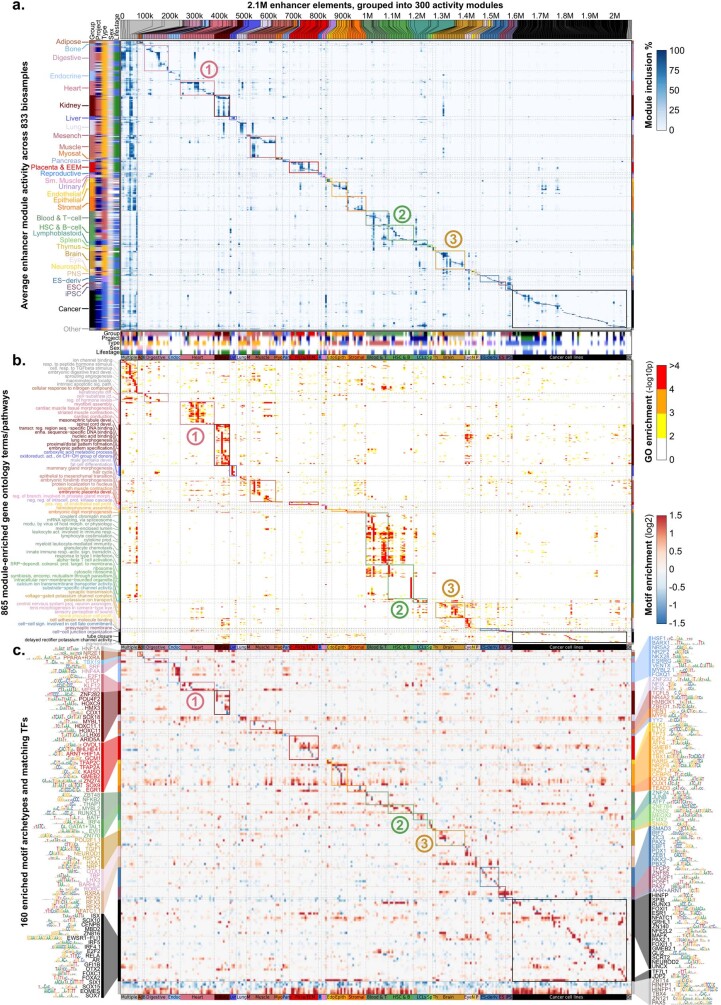
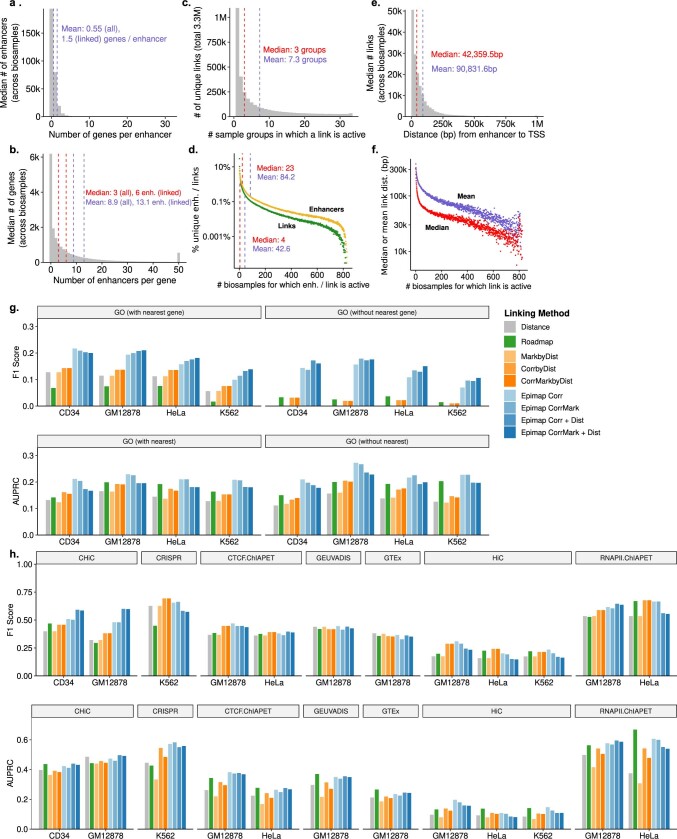
Annotating the molecular basis of human disease remains an unsolved challenge, as 93% of disease loci are non-coding and gene-regulatory annotations are highly incomplete1-3. Here we present EpiMap, a compendium comprising 10,000 epigenomic maps across 800 samples, which we used to define chromatin states, high-resolution enhancers, enhancer modules, upstream regulators and downstream target genes. We used this resource to annotate 30,000 genetic loci that were associated with 540 traits4, predicting trait-relevant tissues, putative causal nucleotide variants in enriched tissue enhancers and candidate tissue-specific target genes for each. We partitioned multifactorial traits into tissue-specific contributing factors with distinct functional enrichments and disease comorbidity patterns, and revealed both single-factor monotropic and multifactor pleiotropic loci. Top-scoring loci frequently had multiple predicted driver variants, converging through multiple enhancers with a common target gene, multiple genes in common tissues, or multiple genes and multiple tissues, indicating extensive pleiotropy. Our results demonstrate the importance of dense, rich, high-resolution epigenomic annotations for the investigation of complex traits.
Conflict of interest statement
The authors declare no competing interests.
Figures








Comment in
-
From polygenic risk scores to integrative epigenomics: the dawn of a new era for cardiovascular precision medicine.Cardiovasc Res. 2021 May 25;117(6):e73-e75. doi: 10.1093/cvr/cvab146. Cardiovasc Res. 2021. PMID: 33914859 Free PMC article. No abstract available.
-
EpiMap: Fine-tuning integrative epigenomics maps to understand complex human regulatory genomic circuitry.Signal Transduct Target Ther. 2021 May 8;6(1):179. doi: 10.1038/s41392-021-00620-5. Signal Transduct Target Ther. 2021. PMID: 33966052 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- HG008155/NH/NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- U24 HG007234/HG/NHGRI NIH HHS/United States
- MH109978/NH/NIH HHS/United States
- MH119509/NH/NIH HHS/United States
- U24 HG009446/HG/NHGRI NIH HHS/United States
- GM087237/NH/NIH HHS/United States
- HG007234/NH/NIH HHS/United States
- R01 MH109978/MH/NIMH NIH HHS/United States
- U01 MH119509/MH/NIMH NIH HHS/United States
- R01 AG058002/AG/NIA NIH HHS/United States
- R01 HG008155/HG/NHGRI NIH HHS/United States
- R35 HG011317/HG/NHGRI NIH HHS/United States
- R01 GM113708/GM/NIGMS NIH HHS/United States
- U01 HG007610/HG/NHGRI NIH HHS/United States
- HG009446/NH/NIH HHS/United States
- HG009088/NH/NIH HHS/United States
- T32 GM087237/GM/NIGMS NIH HHS/United States
- AG058002/NH/NIH HHS/United States
- HG007610/NH/NIH HHS/United States
- GM113708/NH/NIH HHS/United States
- U01 HG009088/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
