

Benchmarking of next and third generation sequencing technologies and their associated algorithms for de novo genome assembly
- PMID: 33537807
- PMCID: PMC7893683
- DOI: 10.3892/mmr.2021.11890
Benchmarking of next and third generation sequencing technologies and their associated algorithms for de novo genome assembly
Abstract
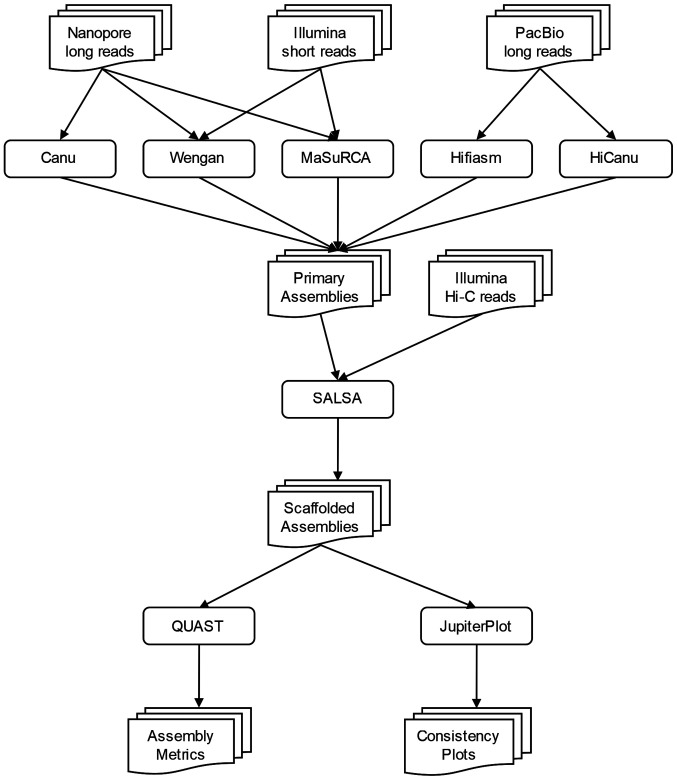
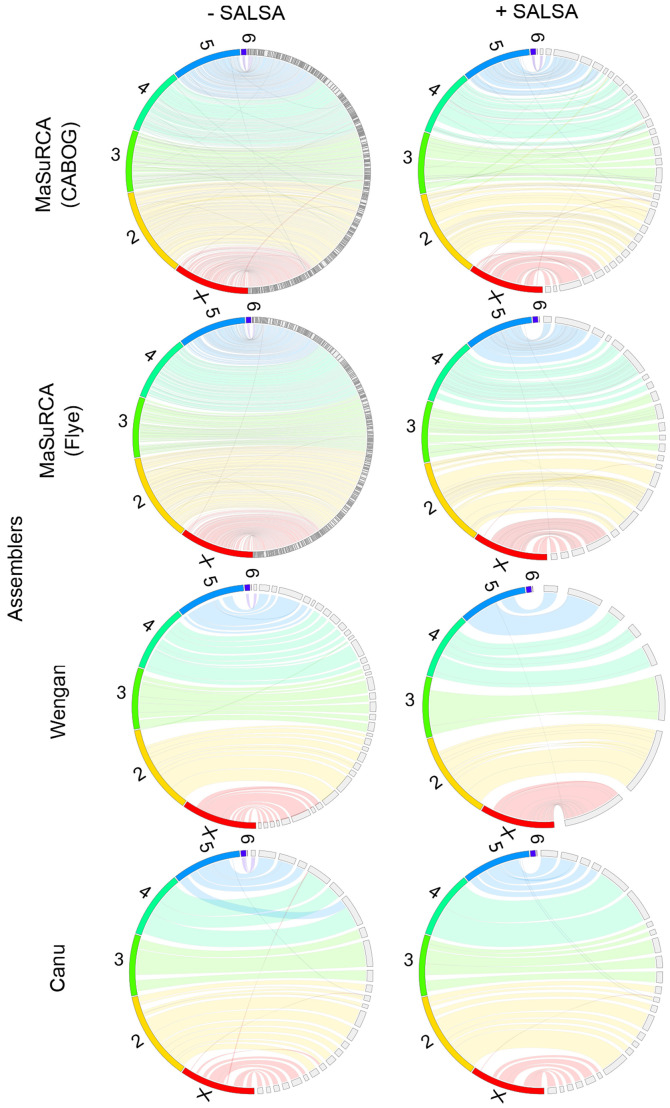
Genome assemblers are computational tools for de novo genome assembly, based on a plenitude of primary sequencing data. The quality of genome assemblies is estimated by their contiguity and the occurrences of misassemblies (duplications, deletions, translocations or inversions). The rapid development of sequencing technologies has enabled the rise of novel de novo genome assembly strategies. The ultimate goal of such strategies is to utilise the features of each sequencing platform in order to address the existing weaknesses of each sequencing type and compose a complete and correct genome map. In the present study, the hybrid strategy, which is based on Illumina short paired‑end reads and Nanopore long reads, was benchmarked using MaSuRCA and Wengan assemblers. Moreover, the long‑read assembly strategy, which is based on Nanopore reads, was benchmarked using Canu or PacBio HiFi reads were benchmarked using Hifiasm and HiCanu. The assemblies were performed on a computational cluster with limited computational resources. Their outputs were evaluated in terms of accuracy and computational performance. PacBio HiFi assembly strategy outperforms the other ones, while Hi‑C scaffolding, which is based on chromatin 3D structure, is required in order to increase continuity, accuracy and completeness when large and complex genomes, such as the human one, are assembled. The use of Hi‑C data is also necessary while using the hybrid assembly strategy. The results revealed that HiFi sequencing enabled the rise of novel algorithms which require less genome coverage than that of the other strategies making the assembly a less computationally demanding task. Taken together, these developments may lead to the democratisation of genome assembly projects which are now approachable by smaller labs with limited technical and financial resources.
Keywords: de novo genome assembly; next generation sequencing; third generation sequencing; genomics; benchmarking; bioinformatics.
Conflict of interest statement
DAS is the Editor-in-Chief for the journal, but had no personal involvement in the reviewing process, or any influence in terms of adjudicating on the final decision, for this article. The other authors declare that they have no competing interests.
Figures



Similar articles
-
Evaluating long-read de novo assembly tools for eukaryotic genomes: insights and considerations.Gigascience. 2022 Dec 28;12:giad100. doi: 10.1093/gigascience/giad100. Epub 2023 Nov 24. Gigascience. 2022. PMID: 38000912 Free PMC article.
-
Benchmarking multi-platform sequencing technologies for human genome assembly.Brief Bioinform. 2023 Sep 20;24(5):bbad300. doi: 10.1093/bib/bbad300. Brief Bioinform. 2023. PMID: 37594299
-
Benchmarking of de novo assembly algorithms for Nanopore data reveals optimal performance of OLC approaches.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):507. doi: 10.1186/s12864-016-2895-8. BMC Genomics. 2016. PMID: 27556636 Free PMC article.
-
The present and future of de novo whole-genome assembly.Brief Bioinform. 2018 Jan 1;19(1):23-40. doi: 10.1093/bib/bbw096. Brief Bioinform. 2018. PMID: 27742661 Review.
-
Chromosome-level hybrid de novo genome assemblies as an attainable option for nonmodel insects.Mol Ecol Resour. 2020 Sep;20(5):1277-1293. doi: 10.1111/1755-0998.13176. Epub 2020 Jun 7. Mol Ecol Resour. 2020. PMID: 32329220 Review.
Cited by
-
A High-Quality Genome Assembly of Striped Catfish (Pangasianodon hypophthalmus) Based on Highly Accurate Long-Read HiFi Sequencing Data.Genes (Basel). 2022 May 22;13(5):923. doi: 10.3390/genes13050923. Genes (Basel). 2022. PMID: 35627308 Free PMC article.
-
A review of the pangenome: how it affects our understanding of genomic variation, selection and breeding in domestic animals?J Anim Sci Biotechnol. 2023 May 5;14(1):73. doi: 10.1186/s40104-023-00860-1. J Anim Sci Biotechnol. 2023. PMID: 37143156 Free PMC article. Review.
-
High-quality Arabidopsis thaliana Genome Assembly with Nanopore and HiFi Long Reads.Genomics Proteomics Bioinformatics. 2022 Feb;20(1):4-13. doi: 10.1016/j.gpb.2021.08.003. Epub 2021 Sep 3. Genomics Proteomics Bioinformatics. 2022. PMID: 34487862 Free PMC article.
-
High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand.Cancers (Basel). 2022 Mar 4;14(5):1325. doi: 10.3390/cancers14051325. Cancers (Basel). 2022. PMID: 35267633 Free PMC article. Review.
-
Evolutionary genomics of three agricultural pest moths reveals rapid evolution of host adaptation and immune-related genes.Gigascience. 2024 Jan 2;13:giad103. doi: 10.1093/gigascience/giad103. Gigascience. 2024. PMID: 38165153 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
