

Moltemplate: A Tool for Coarse-Grained Modeling of Complex Biological Matter and Soft Condensed Matter Physics
- PMID: 33539886
- PMCID: PMC8119336
- DOI: 10.1016/j.jmb.2021.166841
Moltemplate: A Tool for Coarse-Grained Modeling of Complex Biological Matter and Soft Condensed Matter Physics
Abstract
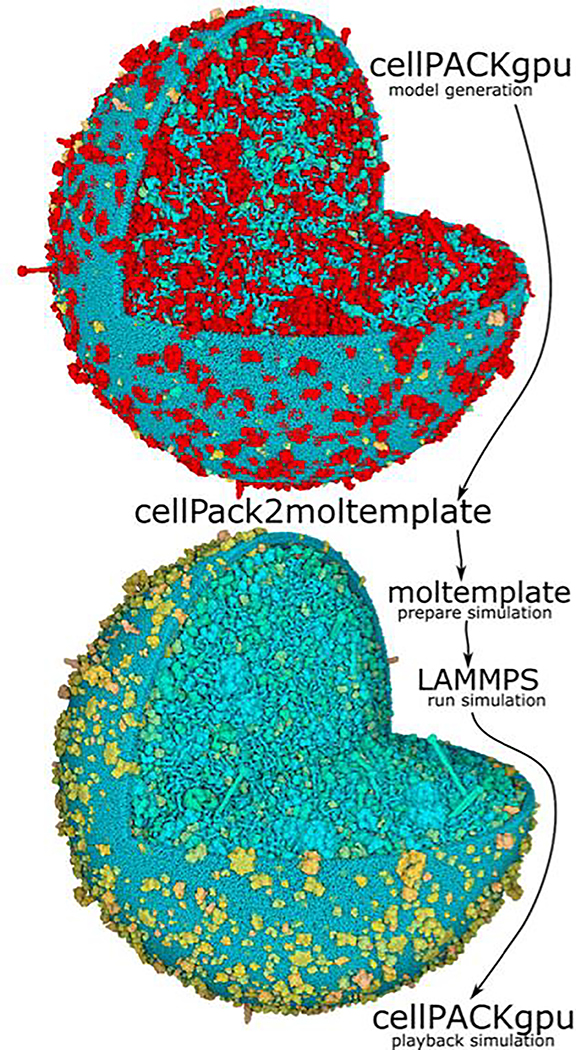
Coarse-grained models have long been considered indispensable tools in the investigation of biomolecular dynamics and assembly. However, the process of simulating such models is arduous because unconventional force fields and particle attributes are often needed, and some systems are not in thermal equilibrium. Although modern molecular dynamics programs are highly adaptable, software designed for preparing all-atom simulations typically makes restrictive assumptions about the nature of the particles and the forces acting on them. Consequently, the use of coarse-grained models has remained challenging. Moltemplate is a file format for storing coarse-grained molecular models and the forces that act on them, as well as a program that converts moltemplate files into input files for LAMMPS, a popular molecular dynamics engine. Moltemplate has broad scope and an emphasis on generality. It accommodates new kinds of forces as they are developed for LAMMPS, making moltemplate a popular tool with thousands of users in computational chemistry, materials science, and structural biology. To demonstrate its wide functionality, we provide examples of using moltemplate to prepare simulations of fluids using many-body forces, coarse-grained organic semiconductors, and the motor-driven supercoiling and condensation of an entire bacterial chromosome.
Keywords: LAMMPS; coarse-grained simulation; molecular dynamics; molecular modeling.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures


References
-
- Senn HM, Thiel W (2009) QM/MM methods for biomolecular systems. Angew Chem Int Ed 48: 1198–1229. - PubMed
-
- Karplus M, McCammon JA (2002) Molecular dynamics simulations of biomolecules. Nat Struct Biol 9: 646–652. - PubMed
-
- Dror RO, Dirks RM, Grossman JP, et al. (2012) Biomolecular simulation: A computational microscope for molecular biology. Annu Rev Biophys 41: 429–452. - PubMed
-
- Marrink SJ, Risselada HJ, Yefimov S, et al. (2007) The MARTINI force field: Coarse grained model for biomolecular simulations. J Phys Chem B 111: 7812–7824. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
