

Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 Å in cells
- PMID: 33542511
- PMCID: PMC7611018
- DOI: 10.1038/s41592-020-01054-7
Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 Å in cells
Abstract
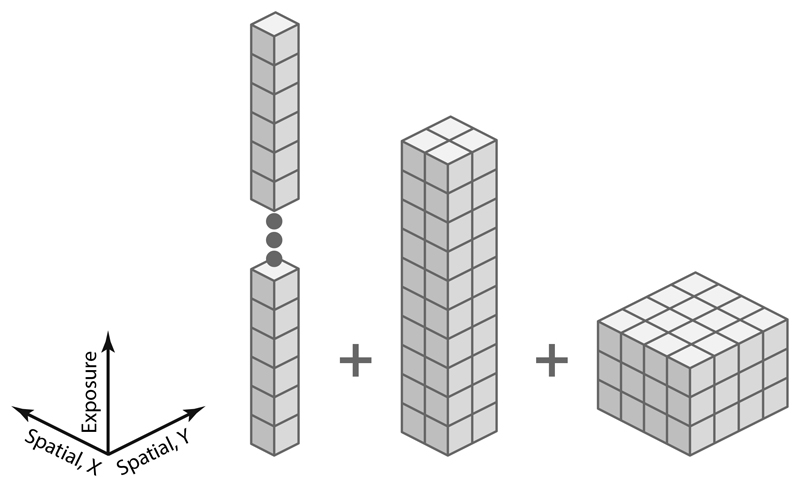
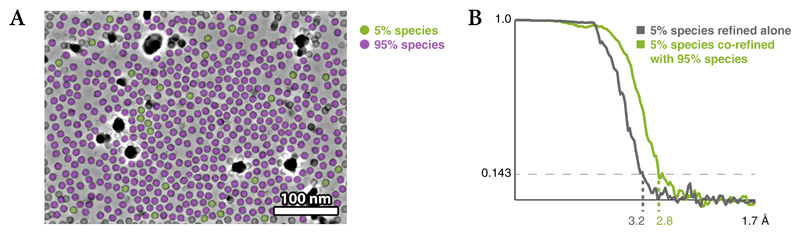
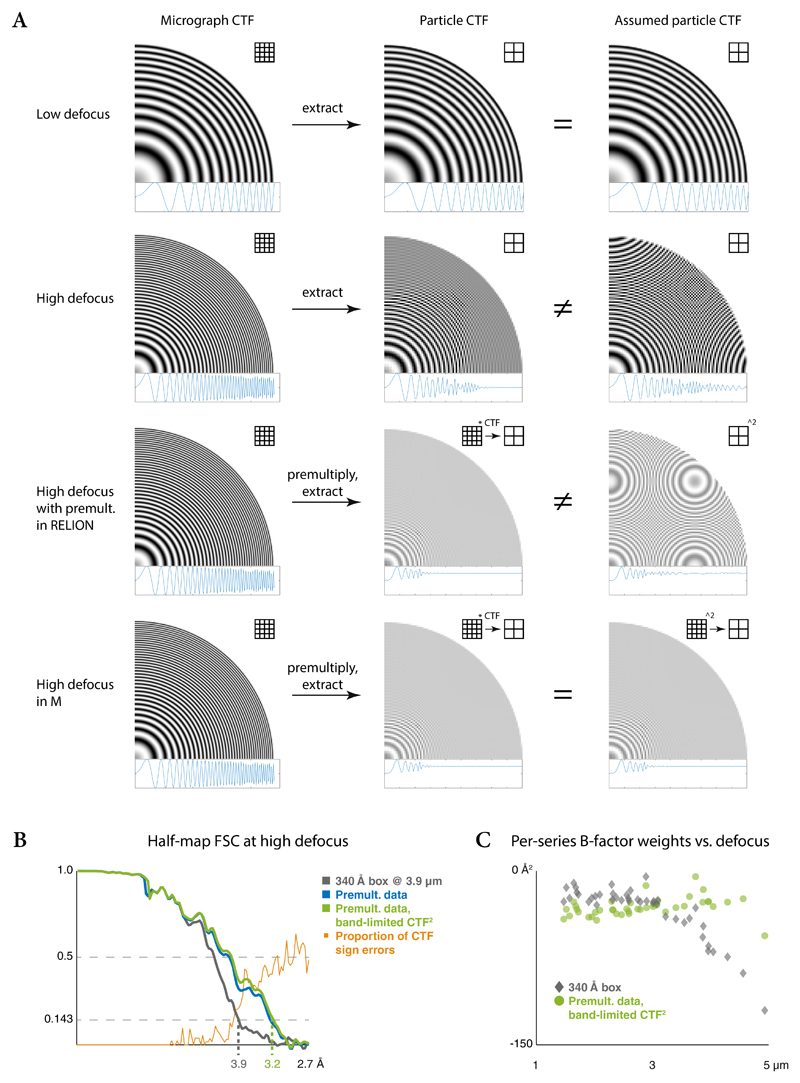
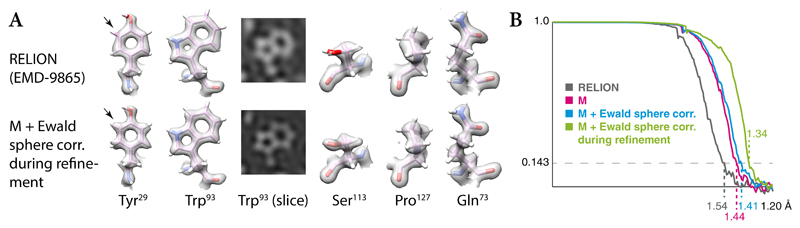
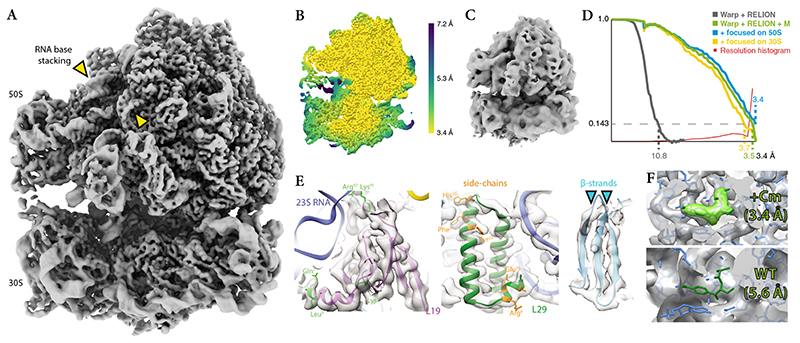
Cryo-electron microscopy (cryo-EM) enables macromolecular structure determination in vitro and inside cells. In addition to aligning individual particles, accurate registration of sample motion and three-dimensional deformation during exposures are crucial for achieving high-resolution reconstructions. Here we describe M, a software tool that establishes a reference-based, multi-particle refinement framework for cryo-EM data and couples a comprehensive spatial deformation model to in silico correction of electron-optical aberrations. M provides a unified optimization framework for both frame-series and tomographic tilt-series data. We show that tilt-series data can provide the same resolution as frame-series data on a purified protein specimen, indicating that the alignment step no longer limits the resolution obtainable from tomographic data. In combination with Warp and RELION, M resolves to residue level a 70S ribosome bound to an antibiotic inside intact bacterial cells. Our work provides a computational tool that facilitates structural biology in cells.
Conflict of interest statement
The authors declare no competing financial or other interests.
Figures

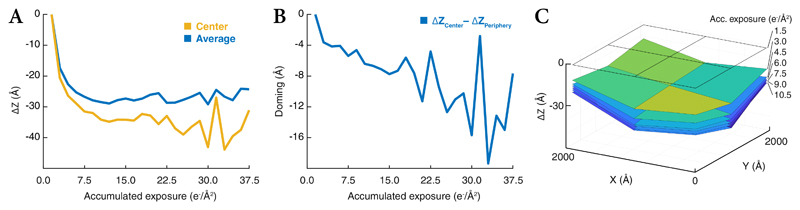
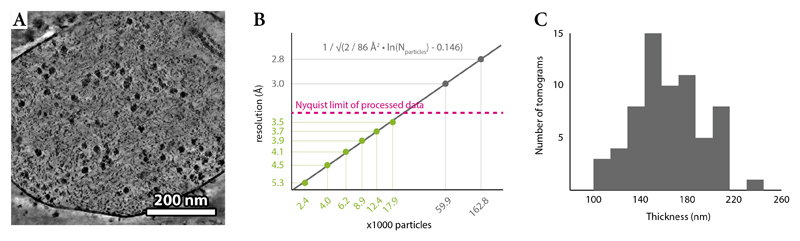
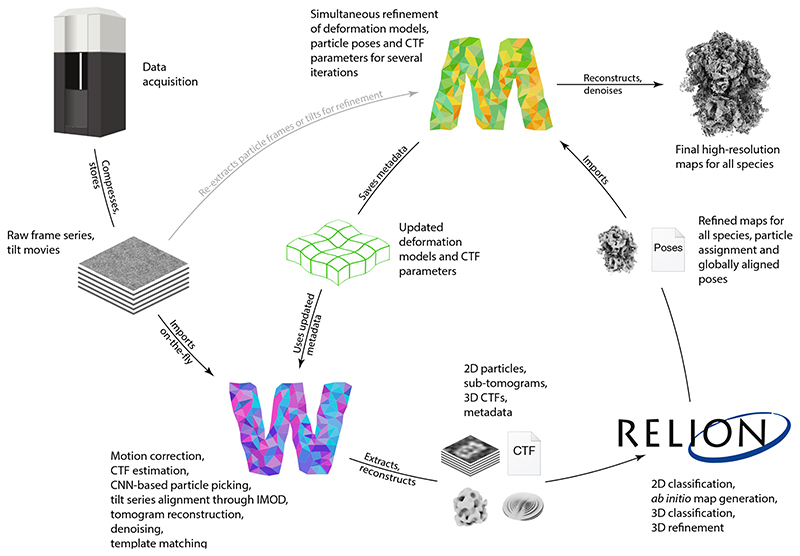
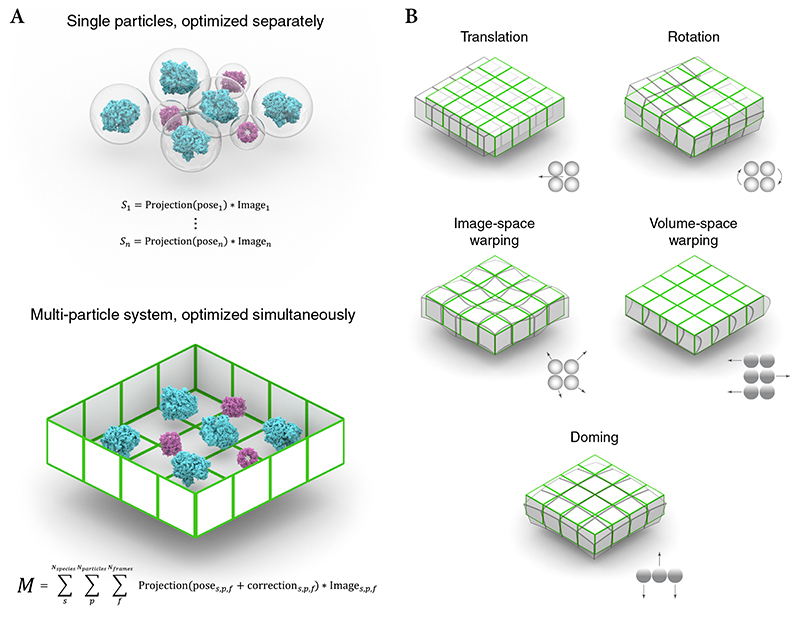
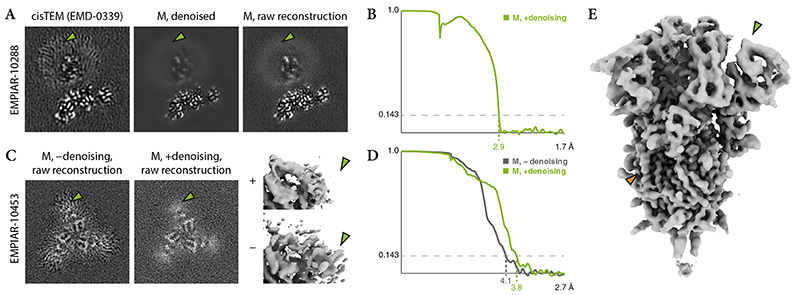
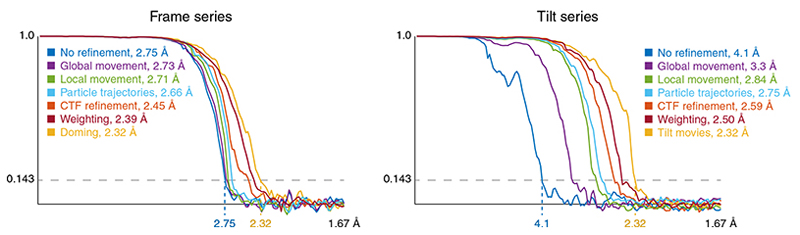
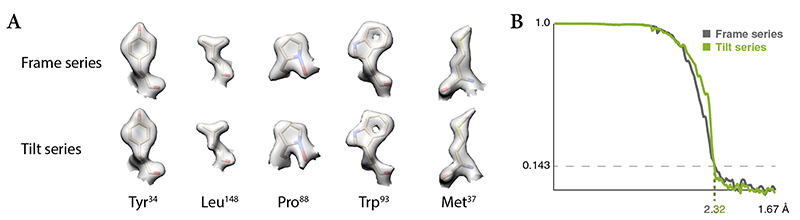
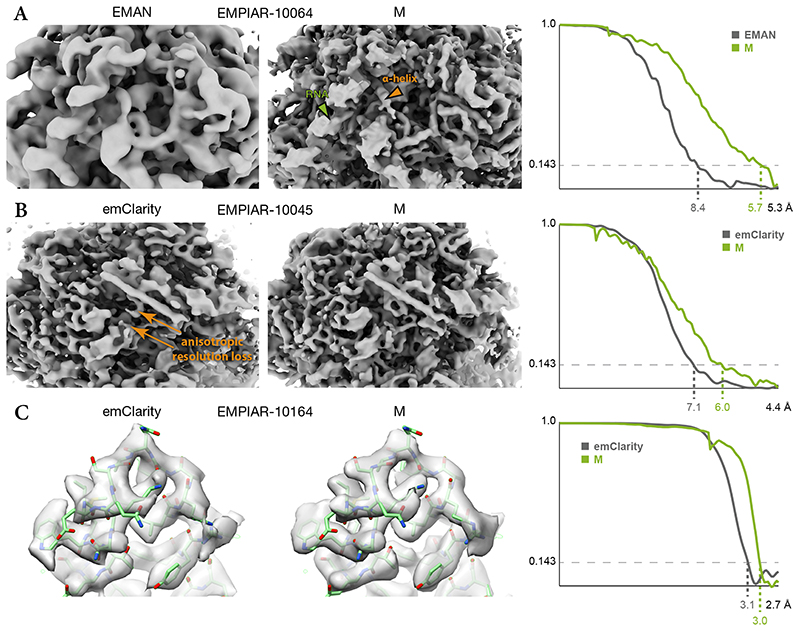
References
-
- Dubochet J, Lepault J, Freeman R, Berriman JA, Homo JC. Electron microscopy of frozen water and aqueous solutions. J Microsc. 1982;128:219–237.
-
- Danev R, Yanagisawa H, Kikkawa M. Cryo-Electron Microscopy Methodology: Current Aspects and Future Directions. Trends in biochemical sciences. 2019;44:837–848. - PubMed
-
- Castaño-Díez D, Kudryashev M, Arheit M, Stahlberg H. Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. Journal of structural biology. 2012;178 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
