

Molecular mechanics and dynamic simulations of well-known Kabuki syndrome-associated KDM6A variants reveal putative mechanisms of dysfunction
- PMID: 33546721
- PMCID: PMC7866879
- DOI: 10.1186/s13023-021-01692-w
Molecular mechanics and dynamic simulations of well-known Kabuki syndrome-associated KDM6A variants reveal putative mechanisms of dysfunction
Erratum in
-
Correction to: Molecular mechanics and dynamic simulations of well-known Kabuki syndrome-associated KDM6A variants reveal putative mechanisms of dysfunction.Orphanet J Rare Dis. 2021 Jun 1;16(1):247. doi: 10.1186/s13023-021-01892-4. Orphanet J Rare Dis. 2021. PMID: 34074320 Free PMC article. No abstract available.
Abstract
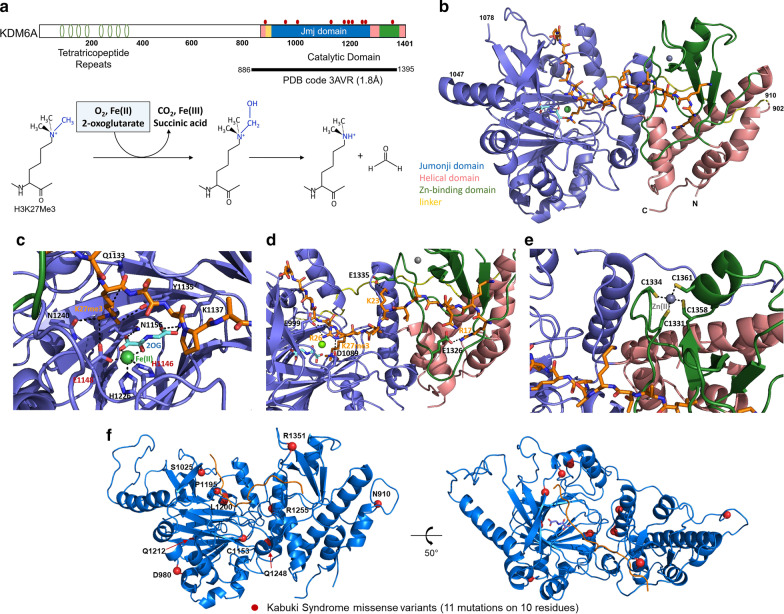
Background: Kabuki syndrome is a genetic disorder that affects several body systems and presents with variations in symptoms and severity. The syndrome is named for a common phenotype of faces resembling stage makeup used in a Japanese traditional theatrical art named kabuki. The most frequent cause of this syndrome is mutations in the H3K4 family of histone methyltransferases while a smaller percentage results from genetic alterations affecting the histone demethylase, KDM6A. Because of the rare presentation of the latter form of the disease, little is known about how missense changes in the KDM6A protein sequence impact protein function.
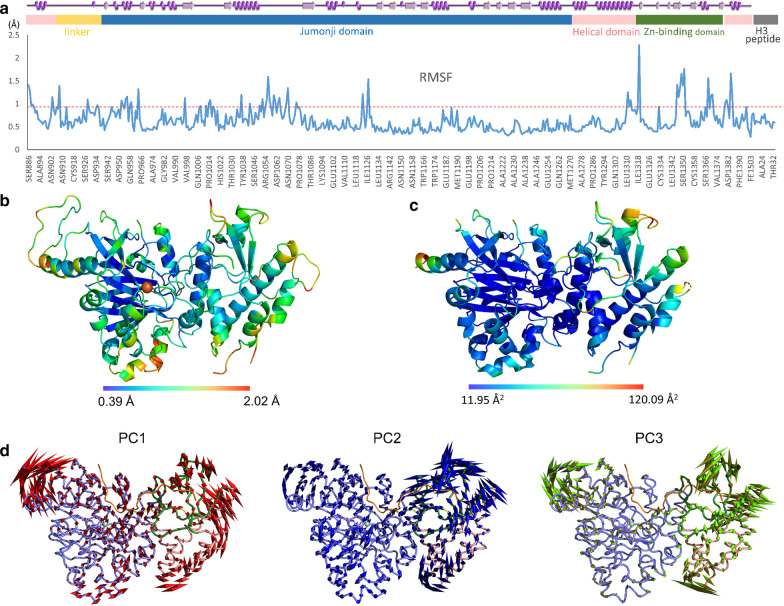
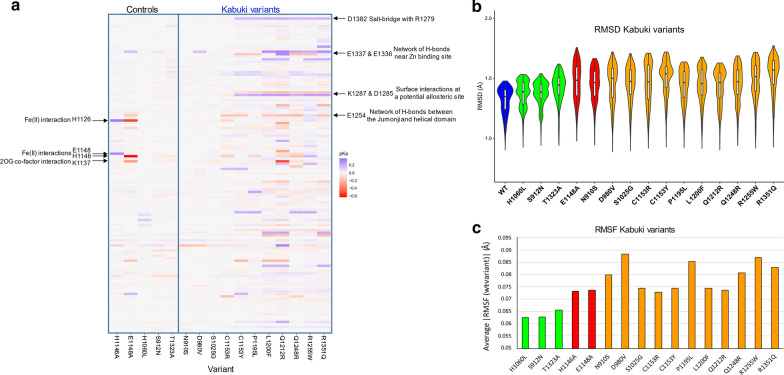
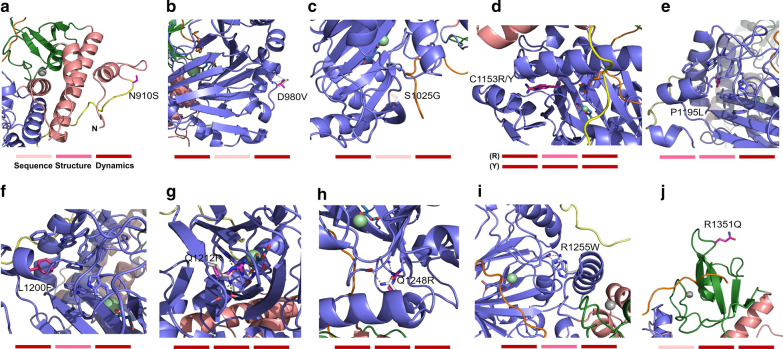
Results: In this study, we use molecular mechanic and molecular dynamic simulations to enhance the annotation and mechanistic interpretation of the potential impact of eleven KDM6A missense variants found in Kabuki syndrome patients. These variants (N910S, D980V, S1025G, C1153R, C1153Y, P1195L, L1200F, Q1212R, Q1248R, R1255W, and R1351Q) are predicted to be pathogenic, likely pathogenic or of uncertain significance by sequence-based analysis. Here, we demonstrate, for the first time, that although Kabuki syndrome missense variants are found outside the functionally critical regions, they could affect overall function by significantly disrupting global and local conformation (C1153R, C1153Y, P1195L, L1200F, Q1212R, Q1248R, R1255W and R1351Q), chemical environment (C1153R, C1153Y, P1195L, L1200F, Q1212R, Q1248R, R1255W and R1351Q), and/or molecular dynamics of the catalytic domain (all variants). In addition, our approaches predict that many mutations, in particular C1153R, could allosterically disrupt the key enzymatic interactions of KDM6A.
Conclusions: Our study demonstrates that the KDM6A Kabuki syndrome variants may impair histone demethylase function through various mechanisms that include altered protein integrity, local environment, molecular interactions and protein dynamics. Molecular dynamics simulations of the wild type and the variants are critical to gain a better understanding of molecular dysfunction. This type of comprehensive structure- and MD-based analyses should help develop improved impact scoring systems to interpret the damaging effects of variants in this protein and other related proteins as well as provide detailed mechanistic insight that is not currently predictable from sequence alone.
Keywords: Epigenetic regulators; Genomic variation; Histone demethylase; KDM6A; Kabuki syndrome; Molecular dynamics; Mutational impact analysis; Protein structure.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Identification of KMT2D and KDM6A variants by targeted sequencing from patients with Kabuki syndrome and other congenital disorders.Gene. 2020 Mar 20;731:144360. doi: 10.1016/j.gene.2020.144360. Epub 2020 Jan 11. Gene. 2020. PMID: 31935506
-
Dissecting KMT2D missense mutations in Kabuki syndrome patients.Hum Mol Genet. 2018 Nov 1;27(21):3651-3668. doi: 10.1093/hmg/ddy241. Hum Mol Genet. 2018. PMID: 30107592 Free PMC article.
-
Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2.Hum Mutat. 2016 Sep;37(9):847-64. doi: 10.1002/humu.23026. Epub 2016 Jul 7. Hum Mutat. 2016. PMID: 27302555
-
A three generation X-linked family with Kabuki syndrome phenotype and a frameshift mutation in KDM6A.Am J Med Genet A. 2014 May;164A(5):1289-92. doi: 10.1002/ajmg.a.36442. Epub 2014 Mar 24. Am J Med Genet A. 2014. PMID: 24664873 Review.
-
De novo exonic deletion of KDM6A in a Chinese girl with Kabuki syndrome: A case report and brief literature review.Am J Med Genet A. 2016 Jun;170(6):1613-21. doi: 10.1002/ajmg.a.37634. Epub 2016 Mar 30. Am J Med Genet A. 2016. PMID: 27028180 Review.
Cited by
-
Case report: A study on the de novo KMT2D variant of Kabuki syndrome with Goodpasture's syndrome by whole exome sequencing.Front Pediatr. 2022 Aug 26;10:933693. doi: 10.3389/fped.2022.933693. eCollection 2022. Front Pediatr. 2022. PMID: 36090579 Free PMC article.
-
Correction to: Molecular mechanics and dynamic simulations of well-known Kabuki syndrome-associated KDM6A variants reveal putative mechanisms of dysfunction.Orphanet J Rare Dis. 2021 Jun 1;16(1):247. doi: 10.1186/s13023-021-01892-4. Orphanet J Rare Dis. 2021. PMID: 34074320 Free PMC article. No abstract available.
-
Structural mechanism of H3K27 demethylation and crosstalk with heterochromatin markers.Mol Cell. 2025 Aug 7;85(15):2869-2884.e6. doi: 10.1016/j.molcel.2025.06.025. Epub 2025 Jul 18. Mol Cell. 2025. PMID: 40683254
-
Pitt-Hopkins syndrome: phenotypic and genotypic description of four unrelated patients and structural analysis of corresponding missense mutations.Neurogenetics. 2021 Jul;22(3):161-169. doi: 10.1007/s10048-021-00651-8. Epub 2021 Jun 14. Neurogenetics. 2021. PMID: 34128147
-
Structural bioinformatics enhances the interpretation of somatic mutations in KDM6A found in human cancers.Comput Struct Biotechnol J. 2022 Apr 28;20:2200-2211. doi: 10.1016/j.csbj.2022.04.028. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35615018 Free PMC article.
References
-
- Kaiwar C, Zimmermann MT, Ferber MJ, Niu Z, Urrutia RA, Klee EW, et al. Novel NR2F1 variants likely disrupt DNA binding: molecular modeling in two cases, review of published cases, genotype-phenotype correlation, and phenotypic expansion of the Bosch-Boonstra-Schaaf optic atrophy syndrome. Cold Spring Harb Mol Case Stud. 2017;3(6):a002162. doi: 10.1101/mcs.a002162. - DOI - PMC - PubMed
-
- Cousin MA, Zimmermann MT, Mathison AJ, Blackburn PR, Boczek NJ, Oliver GR, et al. Functional validation reveals the novel missense V419L variant in TGFBR2 associated with Loeys-Dietz syndrome (LDS) impairs canonical TGF-beta signaling. Cold Spring Harb Mol Case Stud. 2017;3(4):a001727. doi: 10.1101/mcs.a001727. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
