

Targeting oncoproteins with a positive selection assay for protein degraders
- PMID: 33547076
- PMCID: PMC7864573
- DOI: 10.1126/sciadv.abd6263
Targeting oncoproteins with a positive selection assay for protein degraders
Abstract
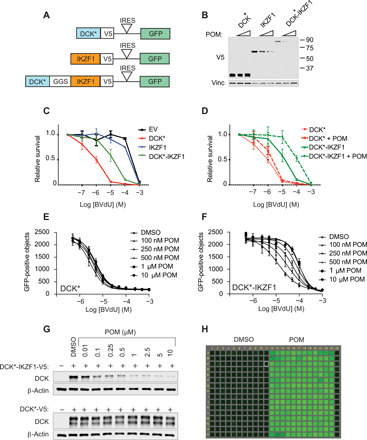
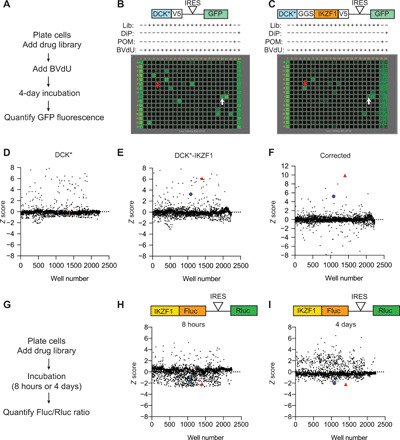
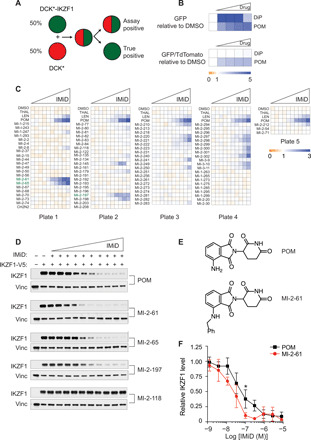
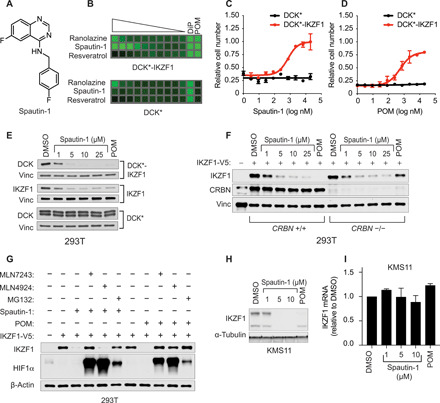
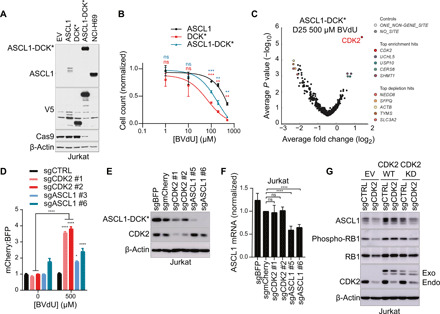
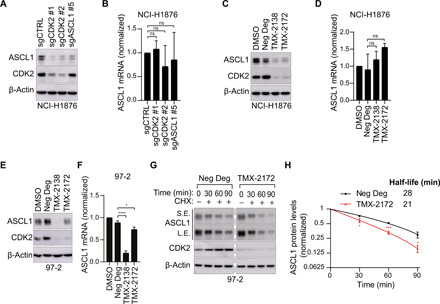
Most intracellular proteins lack hydrophobic pockets suitable for altering their function with drug-like small molecules. Recent studies indicate that some undruggable proteins can be targeted by compounds that can degrade them. For example, thalidomide-like drugs (IMiDs) degrade the critical multiple myeloma transcription factors IKZF1 and IKZF3 by recruiting them to the cereblon E3 ubiquitin ligase. Current loss of signal ("down") assays for identifying degraders often exhibit poor signal-to-noise ratios, narrow dynamic ranges, and false positives from compounds that nonspecifically suppress transcription or translation. Here, we describe a gain of signal ("up") assay for degraders. In arrayed chemical screens, we identified novel IMiD-like IKZF1 degraders and Spautin-1, which, unlike the IMiDs, degrades IKZF1 in a cereblon-independent manner. In a pooled CRISPR-Cas9-based screen, we found that CDK2 regulates the abundance of the ASCL1 oncogenic transcription factor. This methodology should facilitate the identification of drugs that directly or indirectly degrade undruggable proteins.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Krönke J., Udeshi N. D., Narla A., Grauman P., Hurst S. N., Conkey M. M., Svinkina T., Heckl D., Comer E., Li X., Ciarlo C., Hartman E., Munshi N., Schenone M., Schreiber S. L., Carr S. A., Ebert B. L., Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). - PMC - PubMed
-
- Kaelin W. G. Jr., Common pitfalls in preclinical cancer target validation. Nat. Rev. Cancer 17, 441–450 (2017). - PubMed
-
- Gandhi A. K., Kang J., Havens C. G., Conklin T., Ning Y., Wu L., Ito T., Ando H., Waldman M. F., Thakurta A., Klippel A., Handa H., Daniel T. O., Schafer P. H., Chopra R., Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br. J. Haematol. 164, 811–821 (2014). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
