

How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease?
- PMID: 33548031
- PMCID: PMC7886759
- DOI: 10.1007/s12072-020-10121-2
How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease?
Abstract
Background: Non-alcoholic fatty liver disease (NAFLD), characterized as excess lipid accumulation in the liver which is not due to alcohol use, has emerged as one of the major health problems around the world. The dysregulated lipid metabolism creates a lipotoxic environment which promotes the development of NAFLD, especially the progression from simple steatosis (NAFL) to non-alcoholic steatohepatitis (NASH).
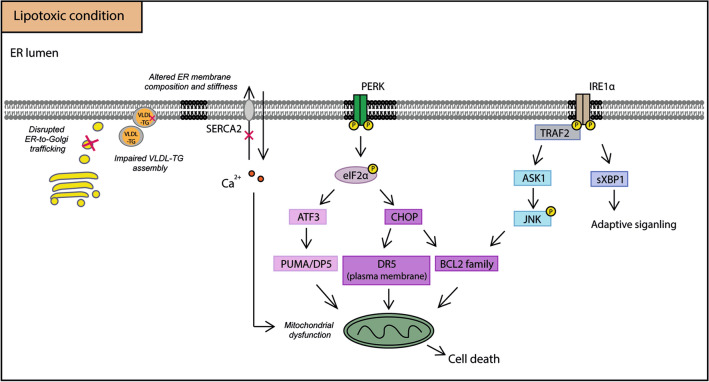
Purposeand aim: This review focuses on the mechanisms of lipid accumulation in the liver, with an emphasis on the metabolic fate of free fatty acids (FFAs) in NAFLD and presents an update on the relevant cellular processes/mechanisms that are involved in lipotoxicity. The changes in the levels of various lipid species that result from the imbalance between lipolysis/lipid uptake/lipogenesis and lipid oxidation/secretion can cause organellar dysfunction, e.g. ER stress, mitochondrial dysfunction, lysosomal dysfunction, JNK activation, secretion of extracellular vesicles (EVs) and aggravate (or be exacerbated by) hypoxia which ultimately lead to cell death. The aim of this review is to provide an overview of how abnormal lipid metabolism leads to lipotoxicity and the cellular mechanisms of lipotoxicity in the context of NAFLD.
Keywords: Cell death; ER stress; Free fatty acids; JNK; Lipid metabolism; Lipotoxicity; MAFLD; Mitochondrial dysfunction; NAFLD; NASH.
Conflict of interest statement
There is no potential conflict of interest.
Figures



References
-
- Eslam M, et al. A new definition for metabolic associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:201. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
