

Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response
- PMID: 33548198
- PMCID: PMC7846228
- DOI: 10.1016/j.chom.2021.01.015
Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response
Abstract
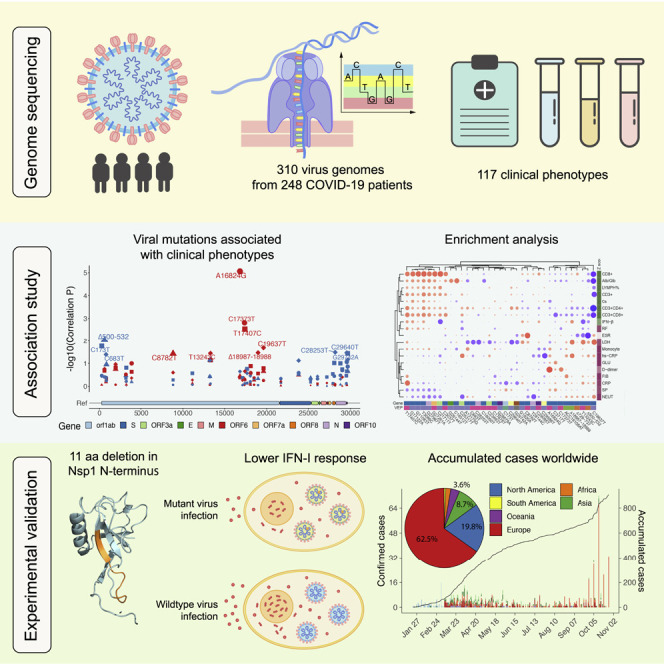
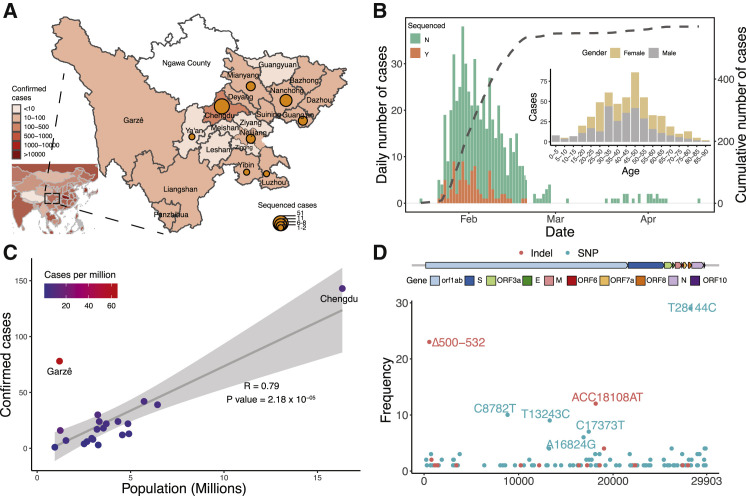
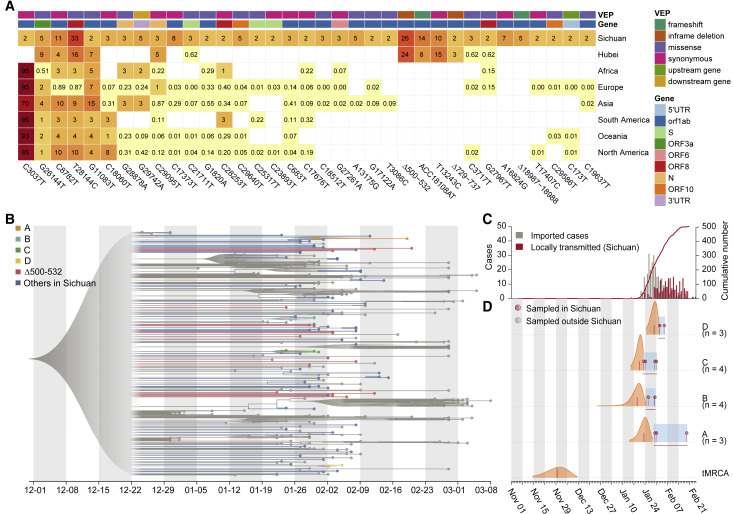
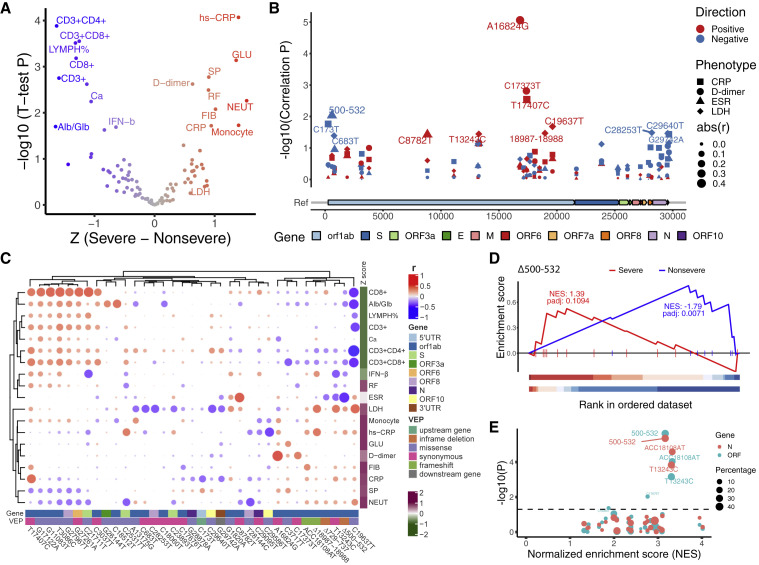
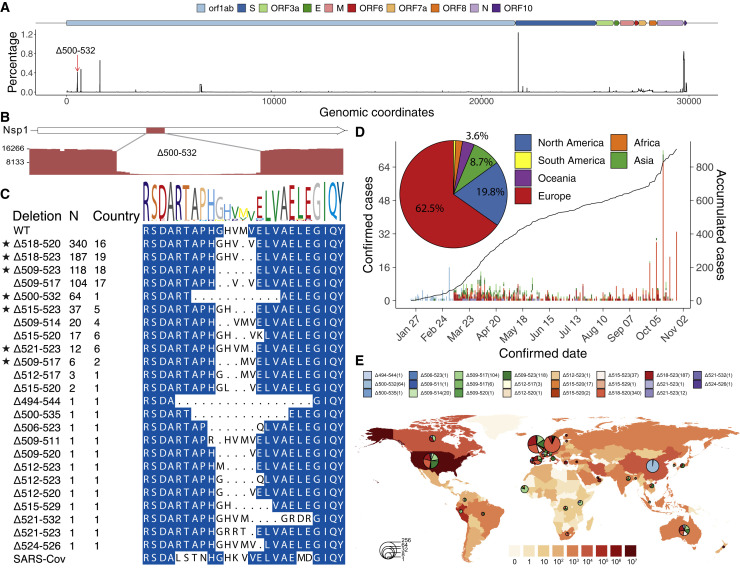
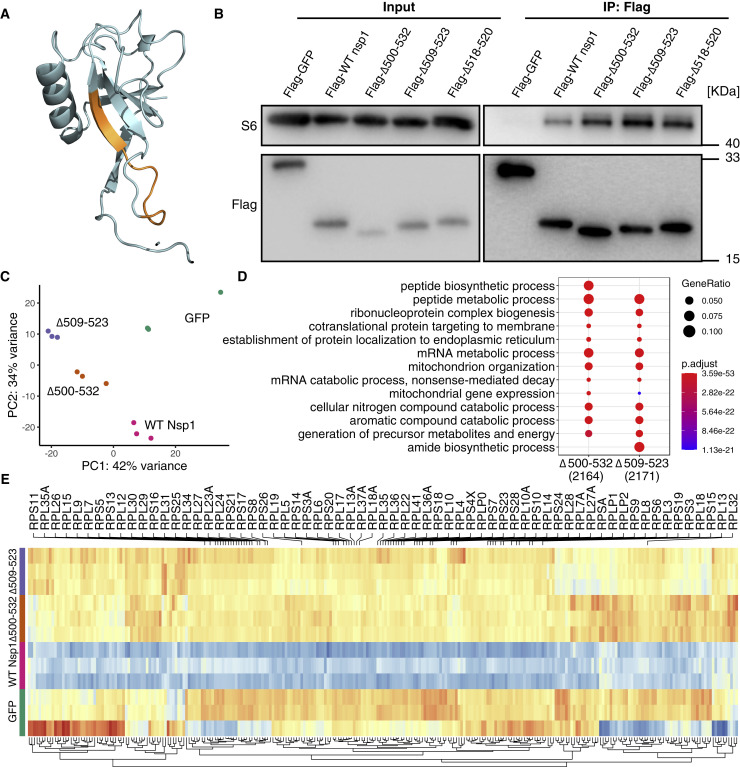
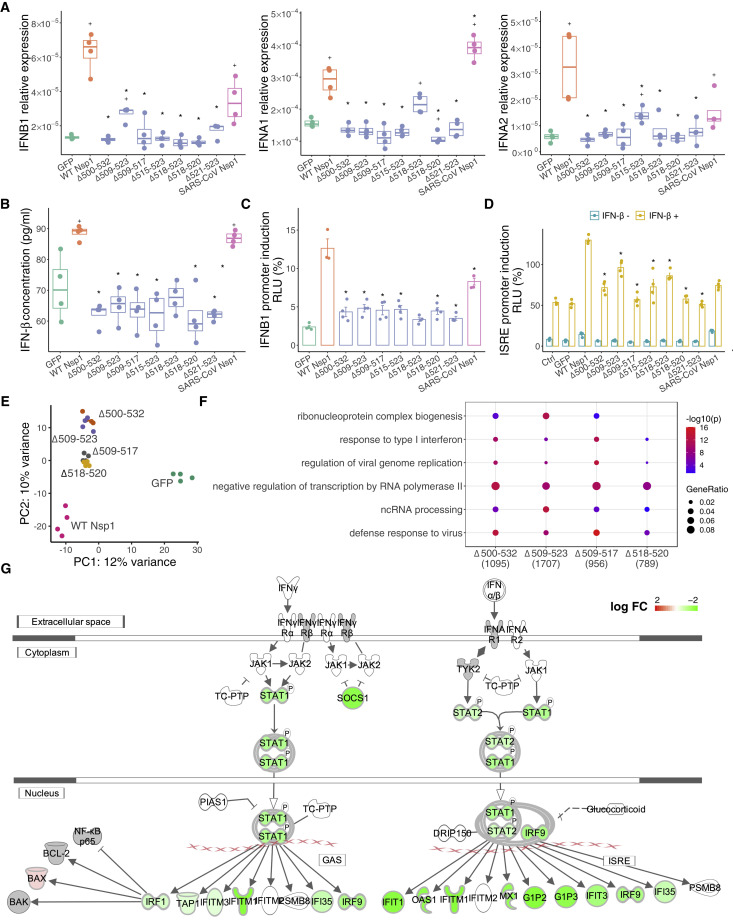
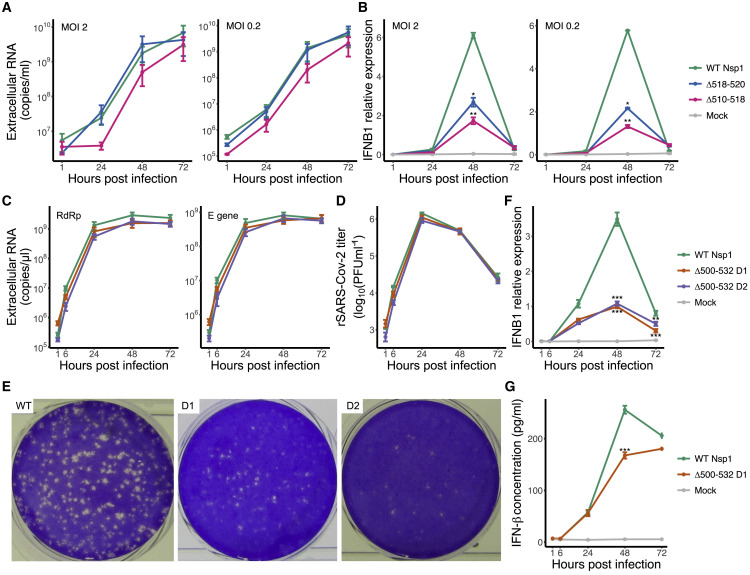
The SARS-CoV-2 virus, the causative agent of COVID-19, is undergoing constant mutation. Here, we utilized an integrative approach combining epidemiology, virus genome sequencing, clinical phenotyping, and experimental validation to locate mutations of clinical importance. We identified 35 recurrent variants, some of which are associated with clinical phenotypes related to severity. One variant, containing a deletion in the Nsp1-coding region (Δ500-532), was found in more than 20% of our sequenced samples and associates with higher RT-PCR cycle thresholds and lower serum IFN-β levels of infected patients. Deletion variants in this locus were found in 37 countries worldwide, and viruses isolated from clinical samples or engineered by reverse genetics with related deletions in Nsp1 also induce lower IFN-β responses in infected Calu-3 cells. Taken together, our virologic surveillance characterizes recurrent genetic diversity and identified mutations in Nsp1 of biological and clinical importance, which collectively may aid molecular diagnostics and drug design.
Keywords: COVID-19; Nsp1; SARS-CoV-2; association; clinical phenotype; deletion; genetic variants; genomic epidemiology; interferon; reverse genetics.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
