

STING, the Endoplasmic Reticulum, and Mitochondria: Is Three a Crowd or a Conversation?
- PMID: 33552072
- PMCID: PMC7858662
- DOI: 10.3389/fimmu.2020.611347
STING, the Endoplasmic Reticulum, and Mitochondria: Is Three a Crowd or a Conversation?
Abstract
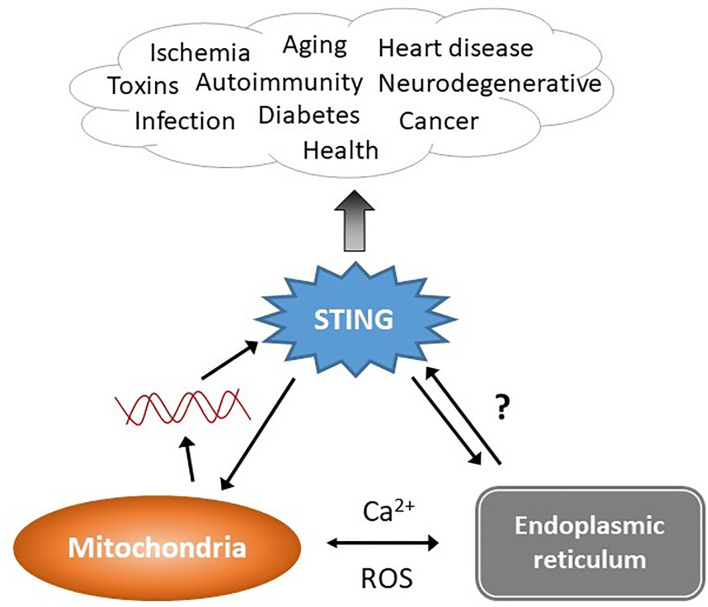
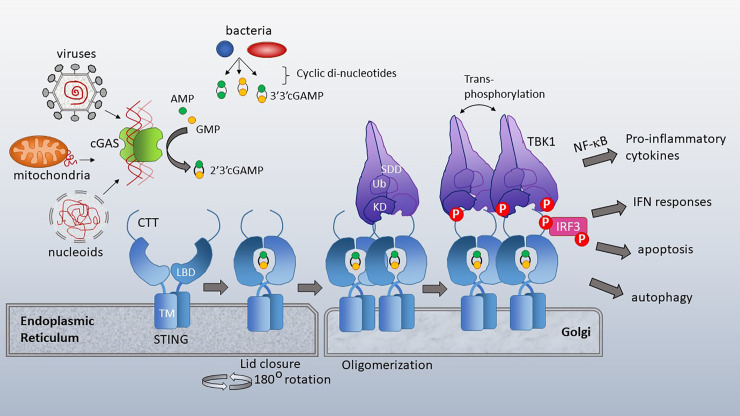
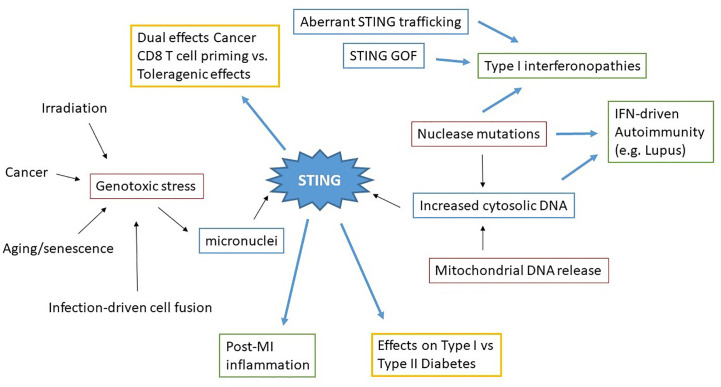
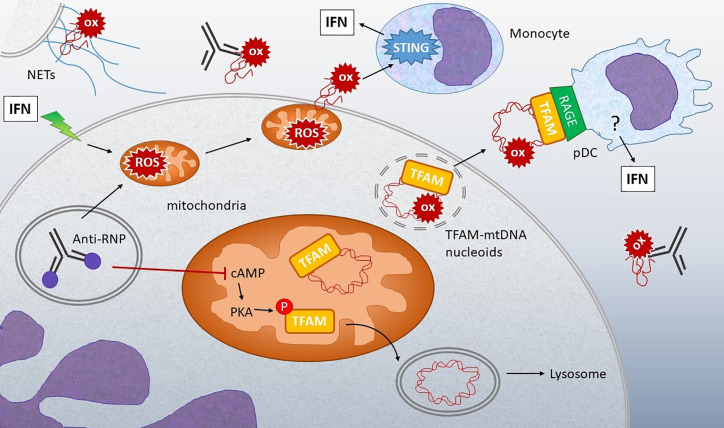
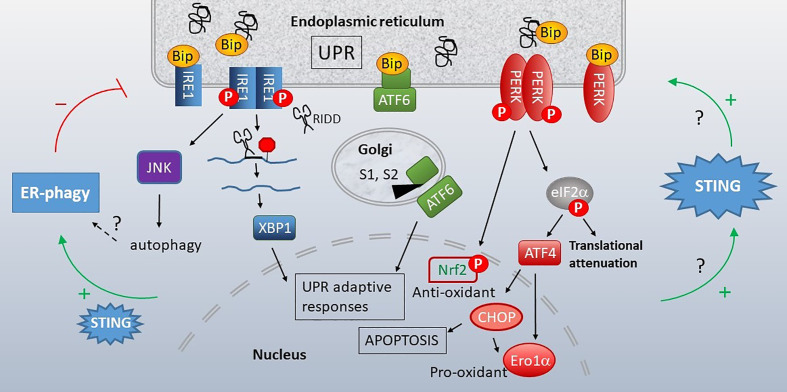
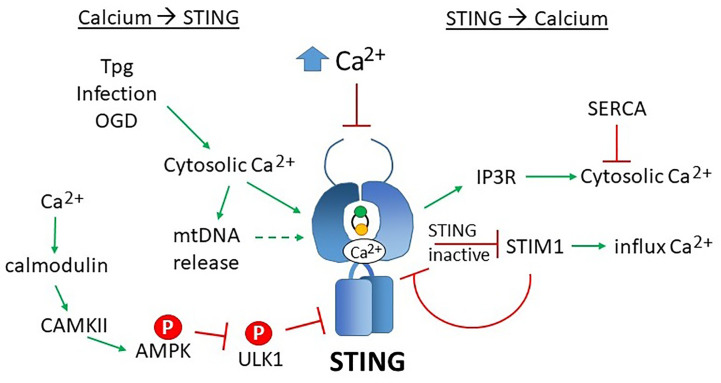
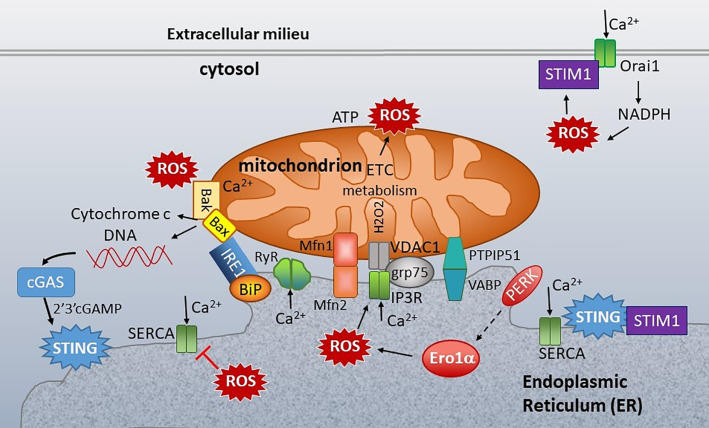
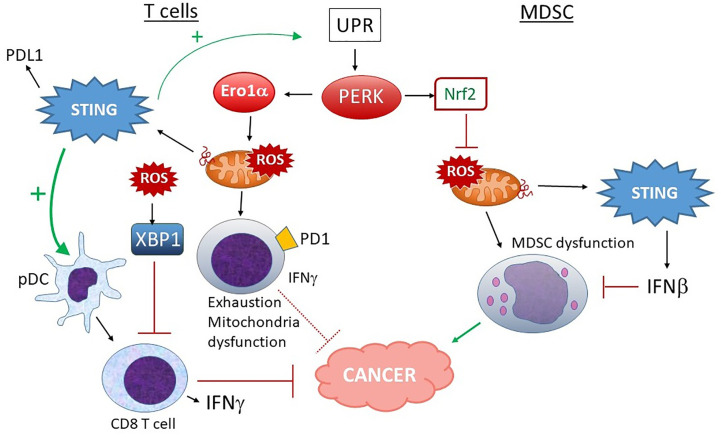
The anti-viral pattern recognition receptor STING and its partnering cytosolic DNA sensor cGAS have been increasingly recognized to respond to self DNA in multiple pathologic settings including cancer and autoimmune disease. Endogenous DNA sources that trigger STING include damaged nuclear DNA in micronuclei and mitochondrial DNA (mtDNA). STING resides in the endoplasmic reticulum (ER), and particularly in the ER-mitochondria associated membranes. This unique location renders STING well poised to respond to intracellular organelle stress. Whereas the pathways linking mtDNA and STING have been addressed recently, the mechanisms governing ER stress and STING interaction remain more opaque. The ER and mitochondria share a close anatomic and functional relationship, with mutual production of, and inter-organelle communication via calcium and reactive oxygen species (ROS). This interdependent relationship has potential to both generate the essential ligands for STING activation and to regulate its activity. Herein, we review the interactions between STING and mitochondria, STING and ER, ER and mitochondria (vis-à-vis calcium and ROS), and the evidence for 3-way communication.
Keywords: STING; cGAS; endoplasmic reticulum; mitochondria; reactive oxygen species; unfolded protein response.
Copyright © 2021 Smith.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials