

[Thrombotic microangiopathy]
- PMID: 33552303
- PMCID: PMC7856846
- DOI: 10.1007/s11560-021-00487-1
[Thrombotic microangiopathy]
Abstract
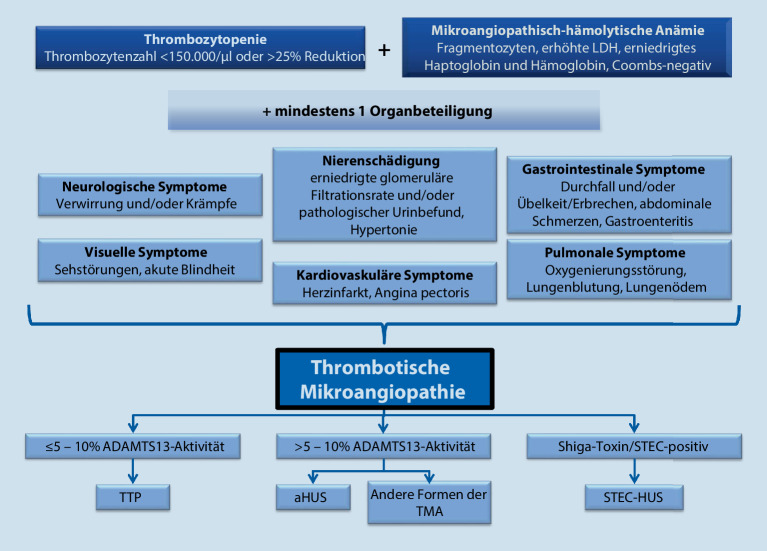
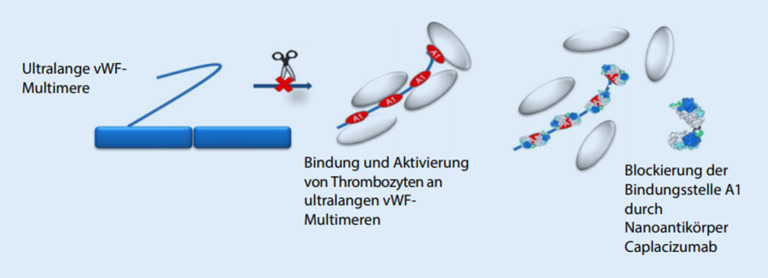
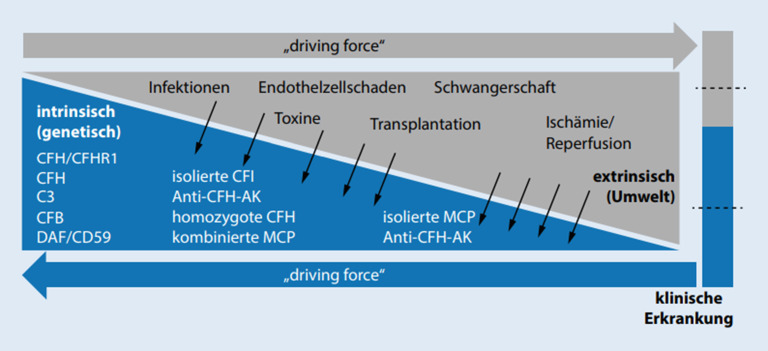
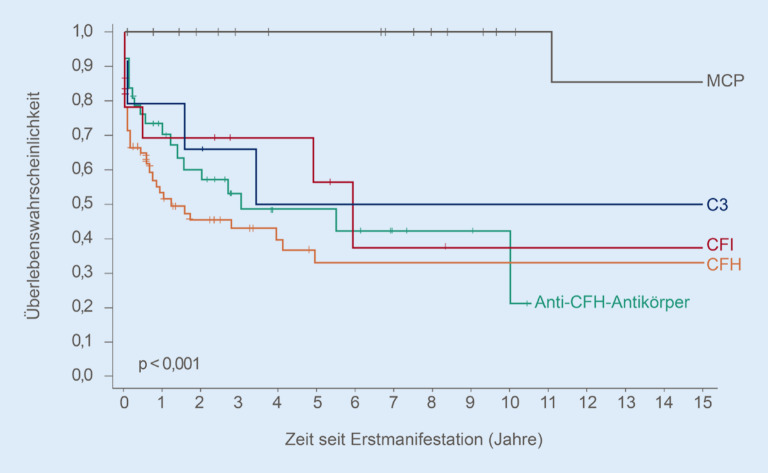
Thrombotic microangiopathy (TMA) is characterized by an endothelium injury-associated formation of platelet clots in arterial and venous microvessels. Concomitant ischemia causes severe organ dysfunction and can be acutely life threatening. The underlying etiology of TMA shows a very heterogeneous disease spectrum. In addition to thrombotic thrombocytopenic purpura, which is characterized by a greatly reduced activity of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), infection-associated classical hemolytic uremic syndrome (HUS) and complement-mediated atypical HUS (aHUS), further very rare diseases or secondary forms can be present. The differential diagnostic classification is pivotal as different treatment approaches are necessary. Initiation of novel specific pharmacotherapy methods has greatly improved the prognosis of TMA.
Die thrombotische Mikroangiopathie (TMA) zeichnet sich durch eine endothelschadenassoziierte Bildung von Plättchenthromben in arteriellen und venösen Mikrogefäßen aus. Die damit einhergehende Ischämie führt zu schwerwiegenden Organdysfunktionen und kann akut lebensbedrohlich sein. Ätiologisch verbirgt sich hinter der TMA ein sehr heterogenes Erkrankungsspektrum. Neben der thrombotisch-thrombozytopenischen Purpura, die durch eine stark reduzierte ADAMTS13(„a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13“)-Aktivität gekennzeichnet ist, dem infektassoziierten klassischen hämolytisch-urämischen Syndrom (HUS) sowie dem komplementvermittelten atypischen HUS (aHUS) können weitere sehr seltene Erkrankungen oder sekundäre Formen vorliegen. Die differenzialdiagnostische Einteilung ist aufgrund unterschiedlicher therapeutischer Ansätze erforderlich. Der Einsatz neuer spezifischer medikamentöser Behandlungsmethoden hat die Prognose der TMA deutlich verbessert.
Keywords: ADAMTS13; Atypical hemolytic uremic syndrome; Complement; Endothelium; Thrombotic thrombocytopenic purpura.
© Springer Medizin Verlag GmbH, ein Teil von Springer Nature 2021.
References
-
- Campistol J, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421–447. - PubMed
-
- Camilleri R, Scully M, Thomas M, et al. A phenotype-genotype correlation of ADAMTS13 mutations in congenital thrombotic thrombocytopenic purpura patients treated in the United Kingdom. J Thromb Haemost. 2012;10(9):1792–1801. - PubMed
-
- Alwan F, Vendramin C, Liesner R, et al. Characterization and treatment of congenital thrombotci thrombocytopenic purpura. Blood. 2019;133(15):1644–1651. - PubMed
-
- Terrell D, Williams L, Vesely S, Lammle B, Hovinga J, George J. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost. 2005;3:1432–1436. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources