

Phylogenomics of Mycobacterium africanum reveals a new lineage and a complex evolutionary history
- PMID: 33555243
- PMCID: PMC8208692
- DOI: 10.1099/mgen.0.000477
Phylogenomics of Mycobacterium africanum reveals a new lineage and a complex evolutionary history
Abstract
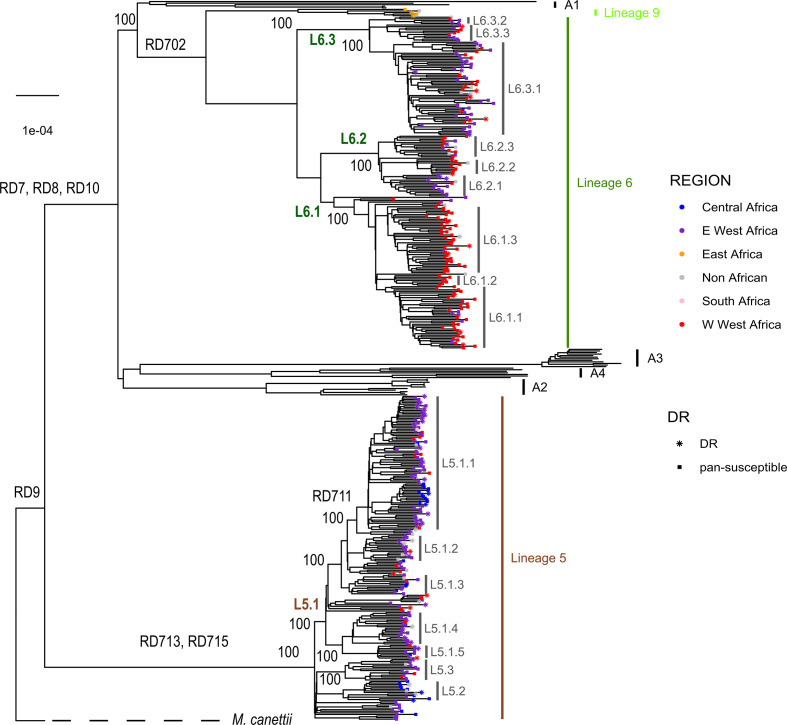
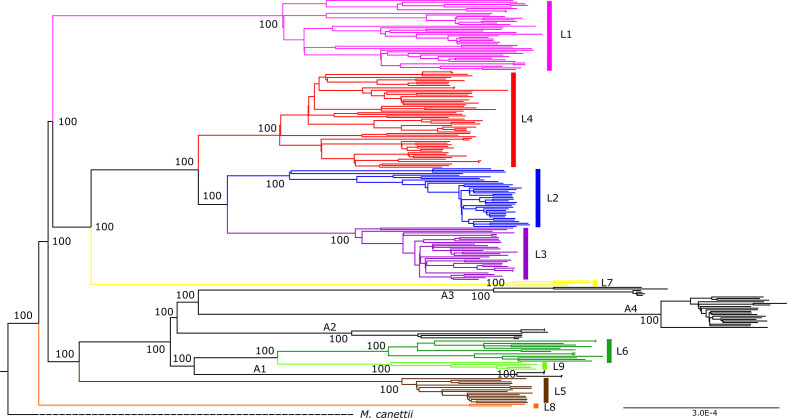
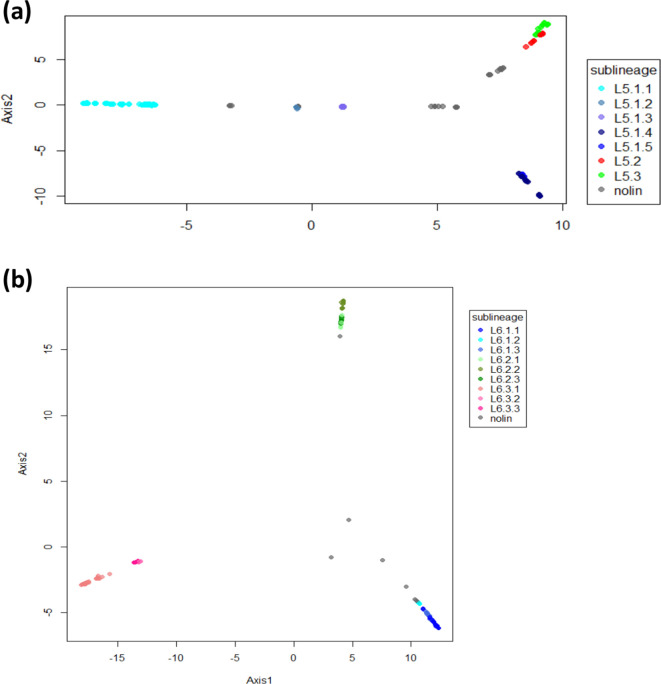
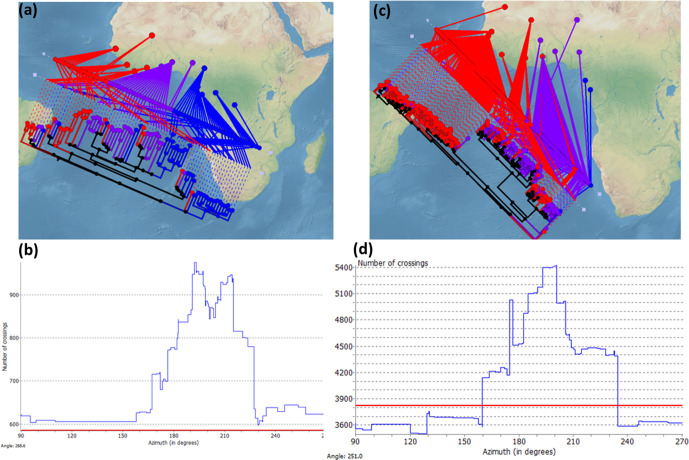
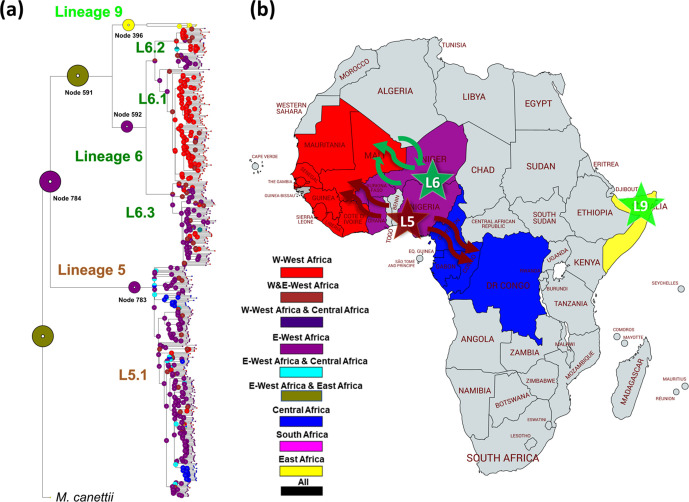
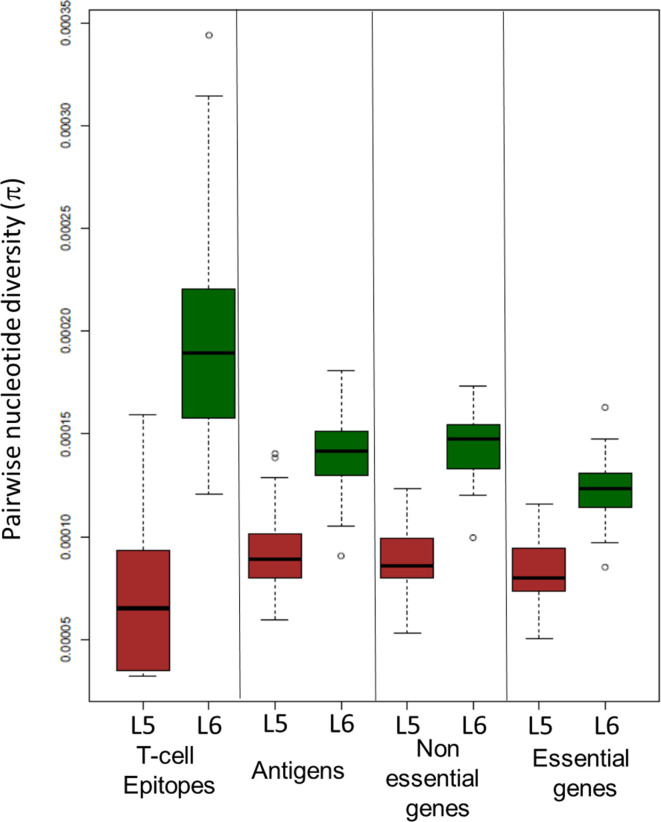
Human tuberculosis (TB) is caused by members of the Mycobacterium tuberculosis complex (MTBC). The MTBC comprises several human-adapted lineages known as M. tuberculosis sensu stricto, as well as two lineages (L5 and L6) traditionally referred to as Mycobacterium africanum. Strains of L5 and L6 are largely limited to West Africa for reasons unknown, and little is known of their genomic diversity, phylogeography and evolution. Here, we analysed the genomes of 350 L5 and 320 L6 strains, isolated from patients from 21 African countries, plus 5 related genomes that had not been classified into any of the known MTBC lineages. Our population genomic and phylogeographical analyses showed that the unclassified genomes belonged to a new group that we propose to name MTBC lineage 9 (L9). While the most likely ancestral distribution of L9 was predicted to be East Africa, the most likely ancestral distribution for both L5 and L6 was the Eastern part of West Africa. Moreover, we found important differences between L5 and L6 strains with respect to their phylogeographical substructure and genetic diversity. Finally, we could not confirm the previous association of drug-resistance markers with lineage and sublineages. Instead, our results indicate that the association of drug resistance with lineage is most likely driven by sample bias or geography. In conclusion, our study sheds new light onto the genomic diversity and evolutionary history of M. africanum, and highlights the need to consider the particularities of each MTBC lineage for understanding the ecology and epidemiology of TB in Africa and globally.
Keywords: Mycobacterium africanum; Mycobacterium tuberculosis; diversity; evolution; genome; mycobacteria.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
-
- WHO Geneva: World Health Organization; 2019. Global Tuberculosis Report 2019.ISBN 978-92-4-156571-4
-
- Riojas MA, McGough KJ, Rider-Riojas CJ, Rastogi N, Hazbón MH. Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycobacterium tuberculosis . Int J Syst Evol Microbiol. 2018;68:324–332. doi: 10.1099/ijsem.0.002507. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical