

The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling
- PMID: 33558590
- PMCID: PMC8080639
- DOI: 10.1038/s12276-021-00560-8
The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling
Abstract
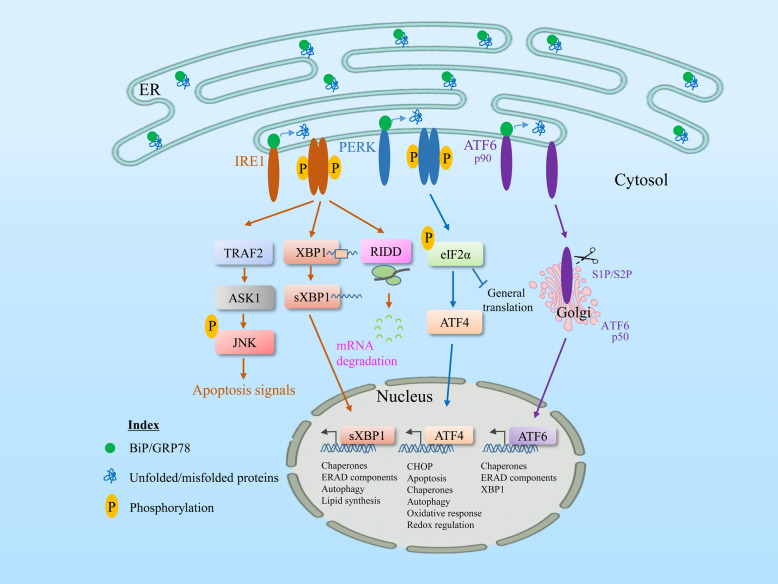
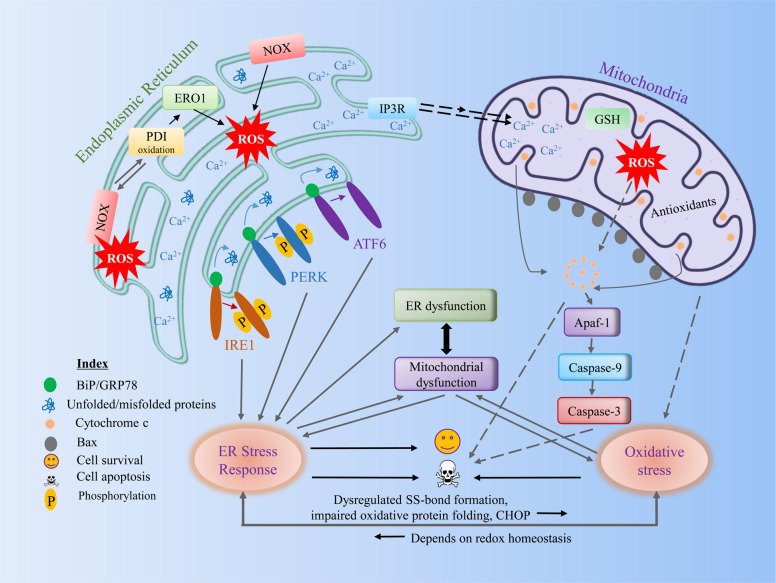
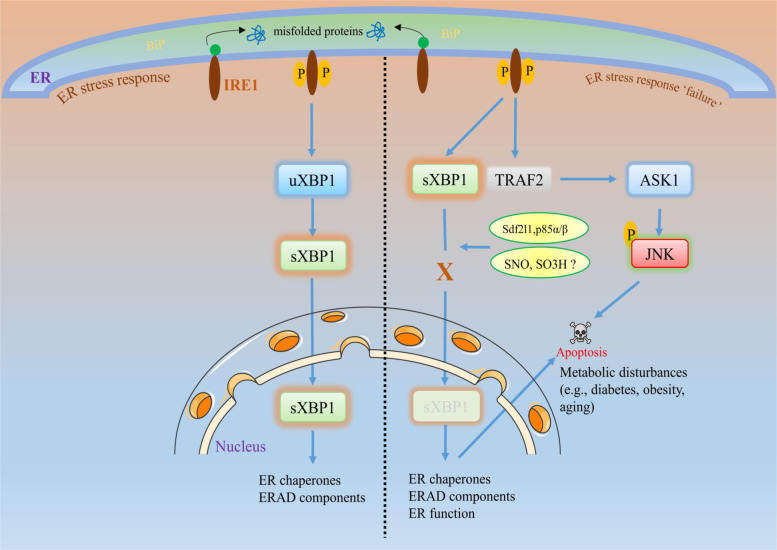
The endoplasmic reticulum (ER) is an essential organelle of eukaryotic cells. Its main functions include protein synthesis, proper protein folding, protein modification, and the transportation of synthesized proteins. Any perturbations in ER function, such as increased demand for protein folding or the accumulation of unfolded or misfolded proteins in the ER lumen, lead to a stress response called the unfolded protein response (UPR). The primary aim of the UPR is to restore cellular homeostasis; however, it triggers apoptotic signaling during prolonged stress. The core mechanisms of the ER stress response, the failure to respond to cellular stress, and the final fate of the cell are not yet clear. Here, we discuss cellular fate during ER stress, cross talk between the ER and mitochondria and its significance, and conditions that can trigger ER stress response failure. We also describe how the redox environment affects the ER stress response, and vice versa, and the aftermath of the ER stress response, integrating a discussion on redox imbalance-induced ER stress response failure progressing to cell death and dynamic pathophysiological changes.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
