

Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex
- PMID: 33558695
- PMCID: PMC8095368
- DOI: 10.1038/s41593-020-00787-0
Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex
Erratum in
-
Publisher Correction: Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex.Nat Neurosci. 2021 Apr;24(4):612. doi: 10.1038/s41593-021-00817-5. Nat Neurosci. 2021. PMID: 33603232 No abstract available.
Abstract
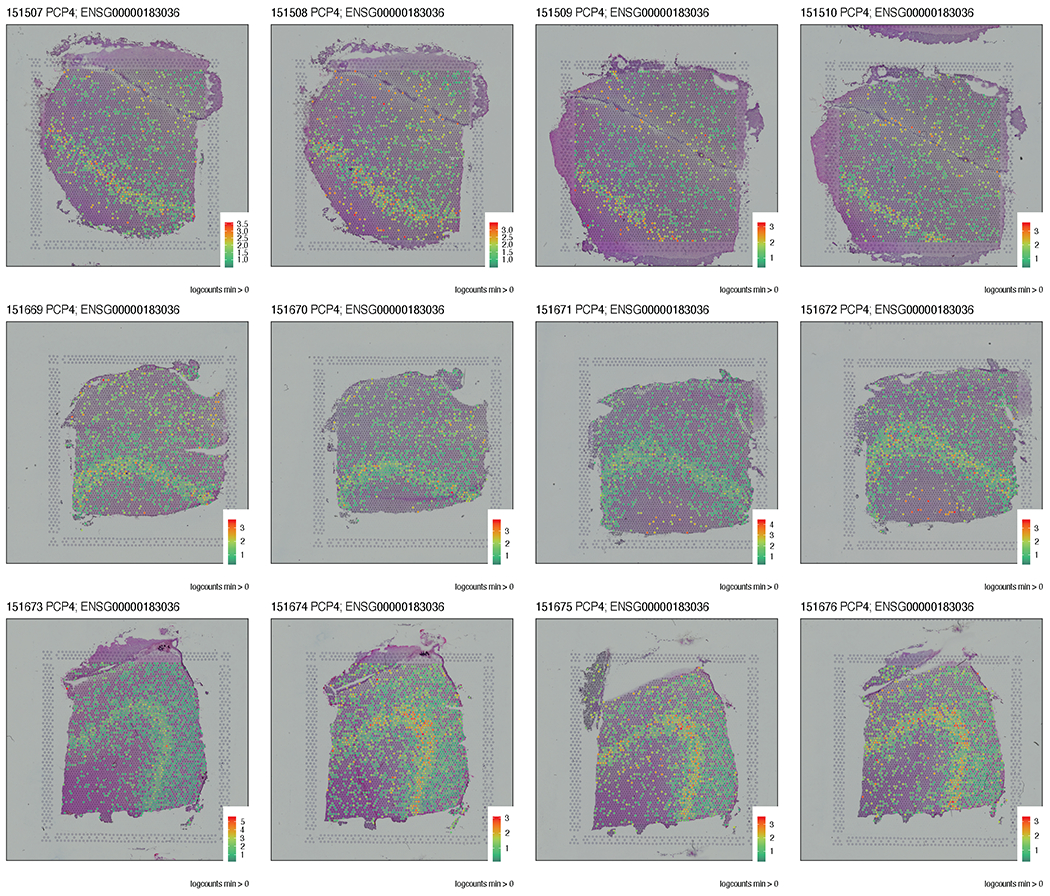
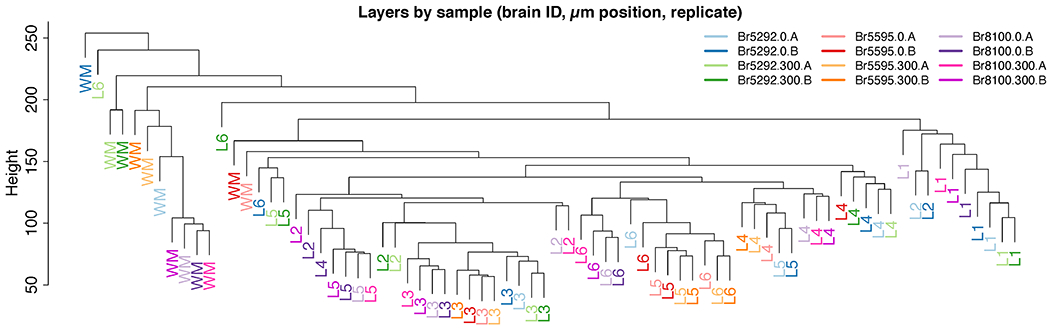
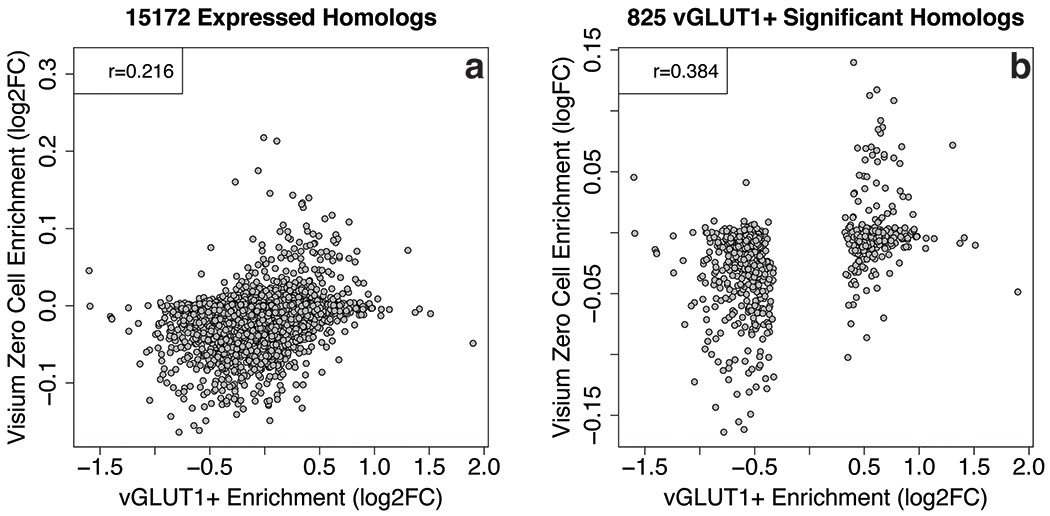
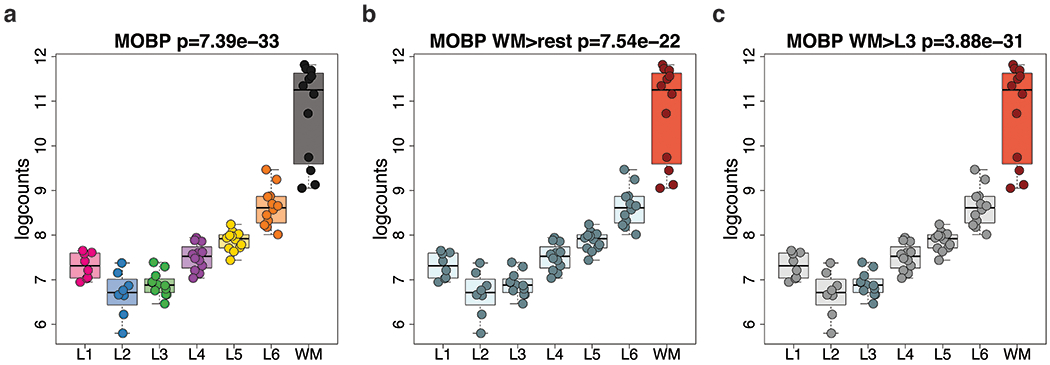
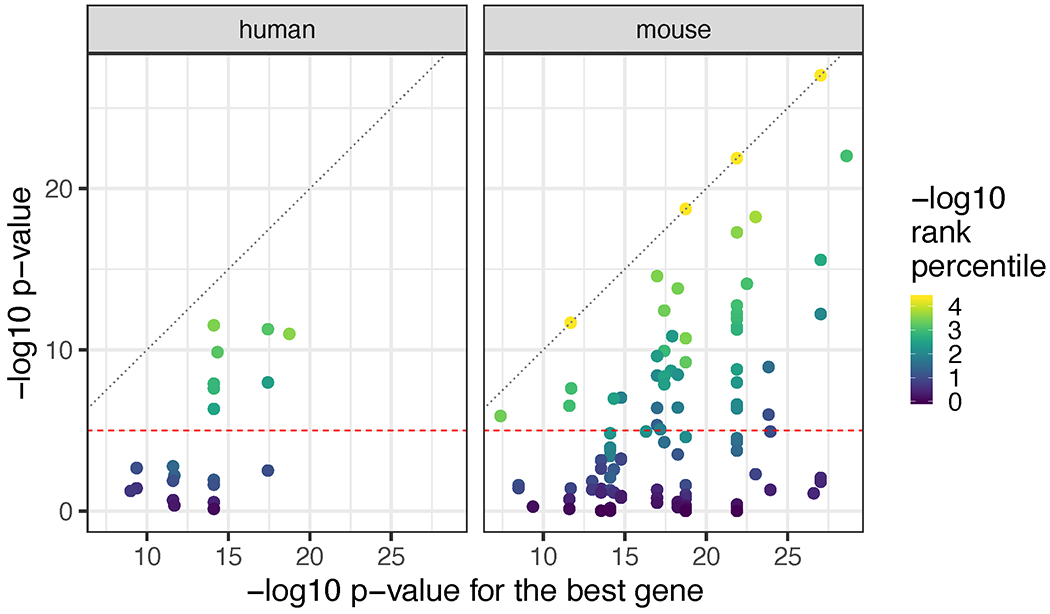
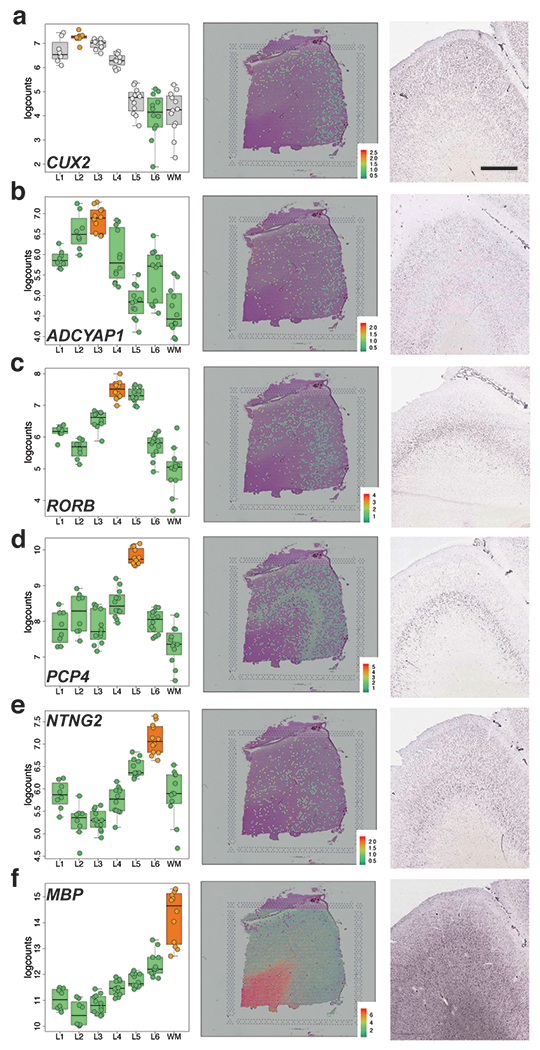
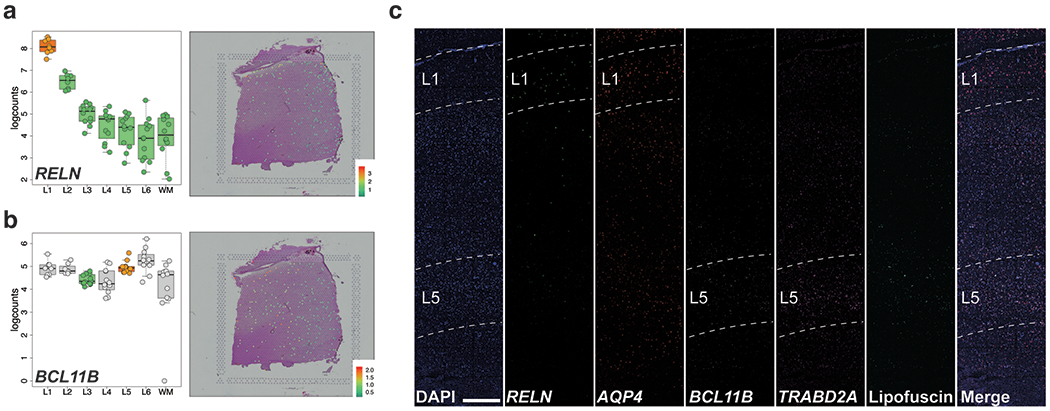
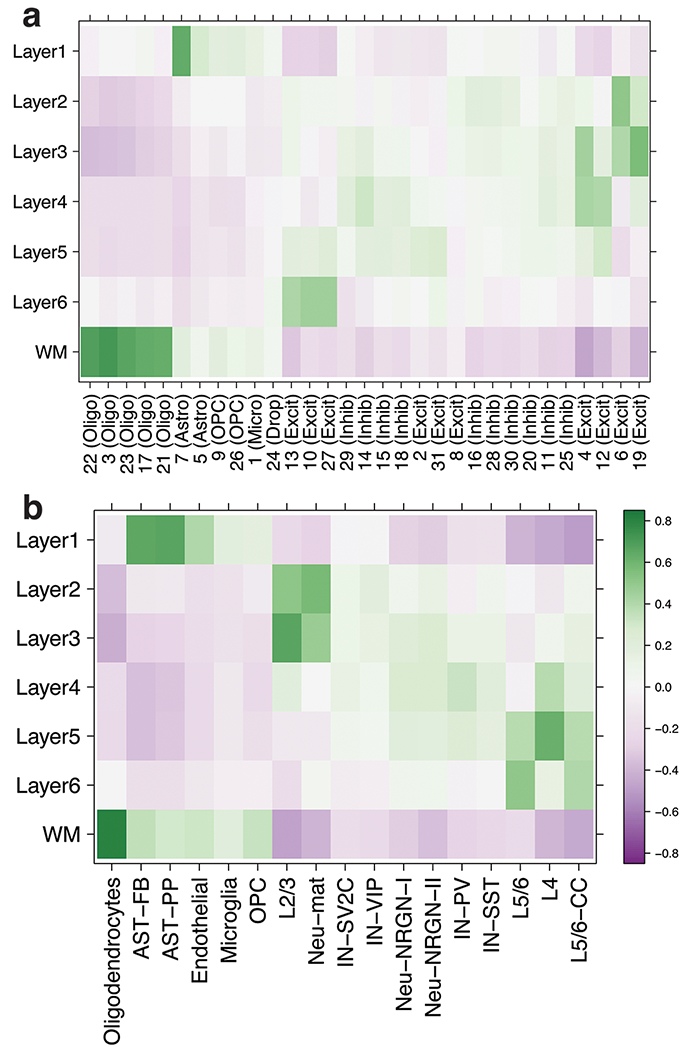
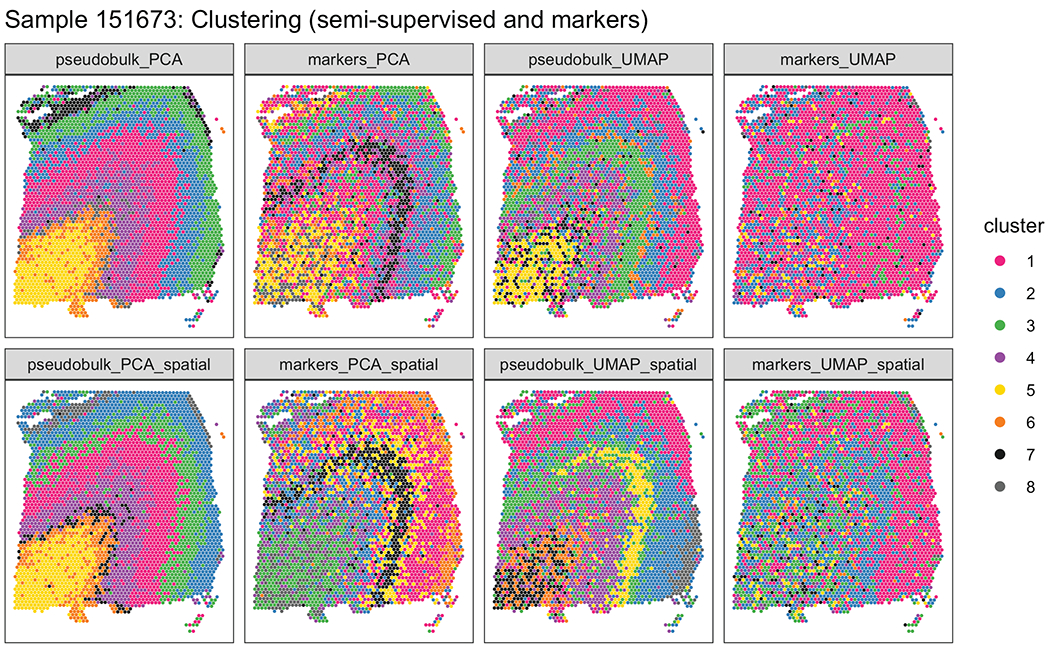
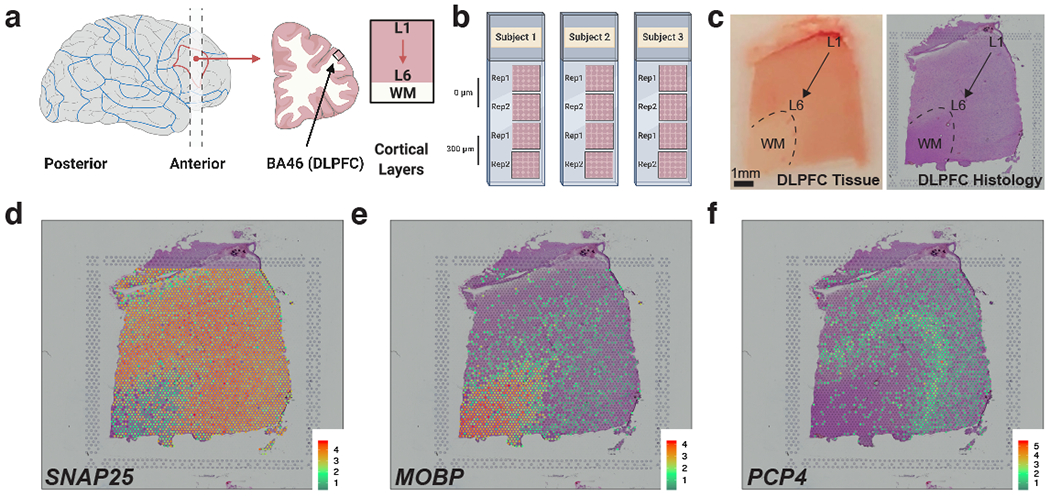
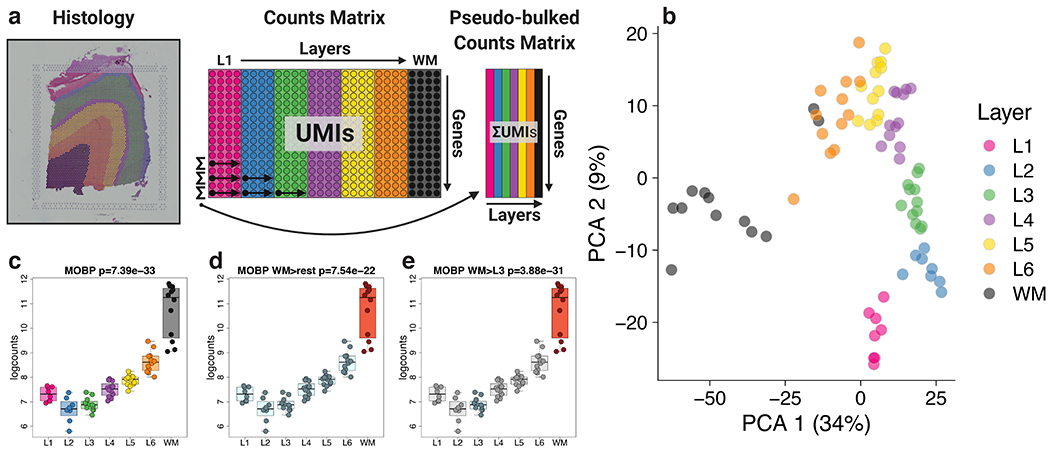
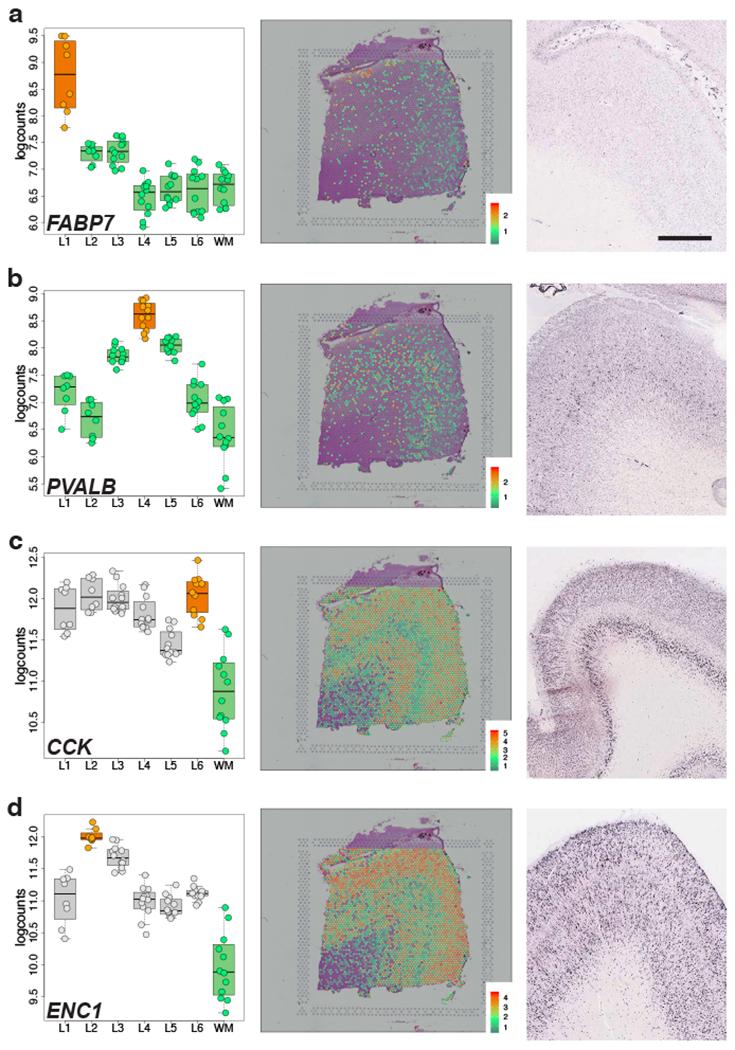
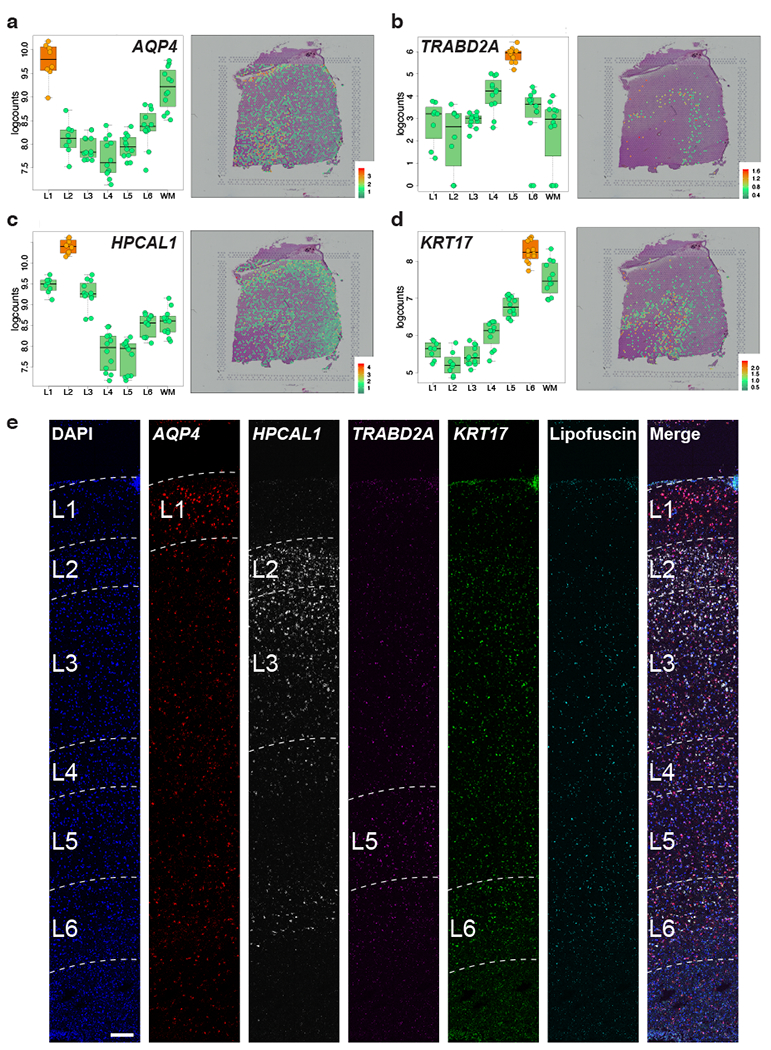
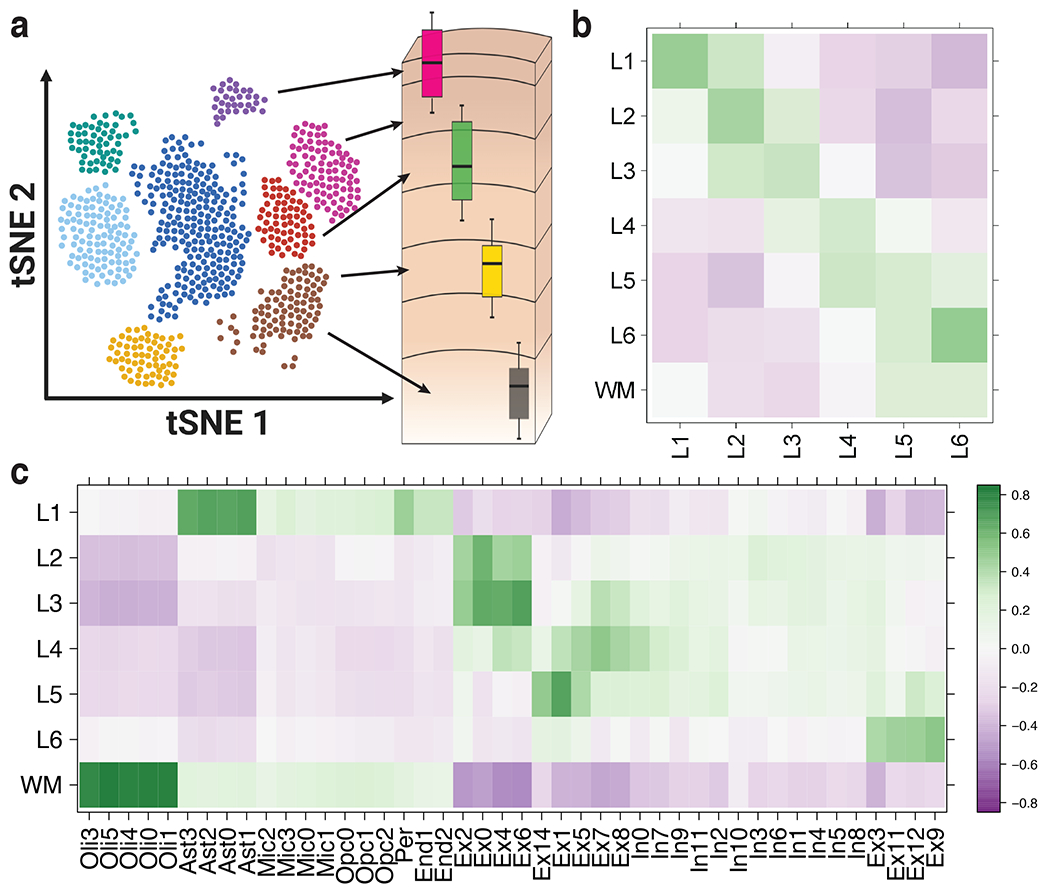
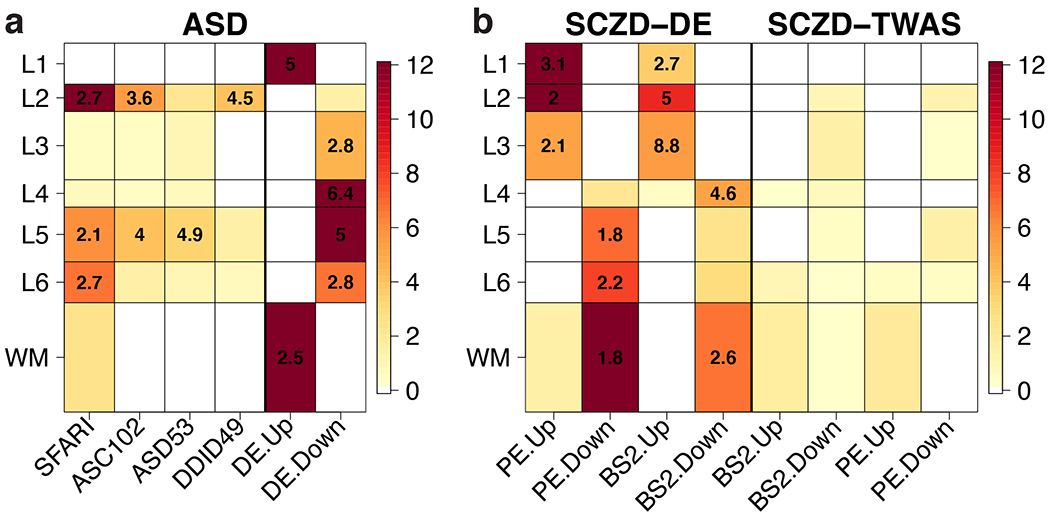
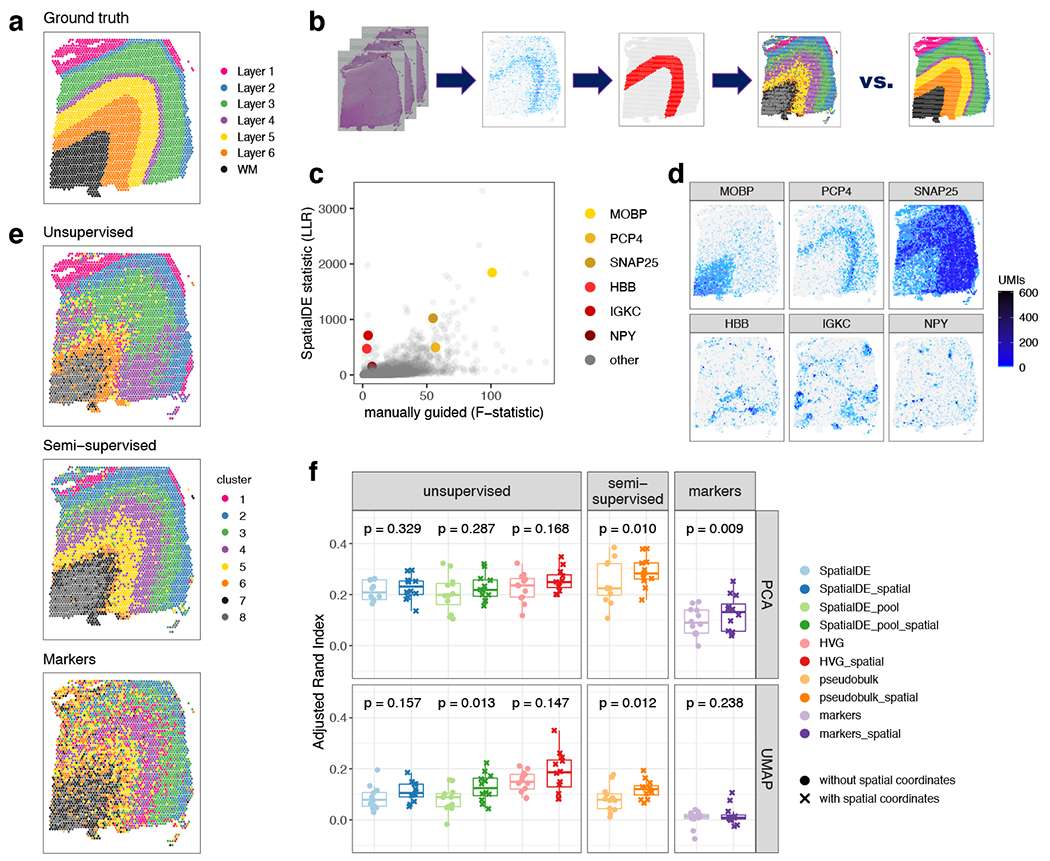
We used the 10x Genomics Visium platform to define the spatial topography of gene expression in the six-layered human dorsolateral prefrontal cortex. We identified extensive layer-enriched expression signatures and refined associations to previous laminar markers. We overlaid our laminar expression signatures on large-scale single nucleus RNA-sequencing data, enhancing spatial annotation of expression-driven clusters. By integrating neuropsychiatric disorder gene sets, we showed differential layer-enriched expression of genes associated with schizophrenia and autism spectrum disorder, highlighting the clinical relevance of spatially defined expression. We then developed a data-driven framework to define unsupervised clusters in spatial transcriptomics data, which can be applied to other tissues or brain regions in which morphological architecture is not as well defined as cortical laminae. Last, we created a web application for the scientific community to explore these raw and summarized data to augment ongoing neuroscience and spatial transcriptomics research ( http://research.libd.org/spatialLIBD ).
Conflict of interest statement
Competing Interests Statement
C.U., S.R.W., J.C., Y.Y., and N.R. are employees of 10x Genomics. All other authors have no conflicts of interest to declare.
Figures

References
-
- DeFelipe J & Fariñas I The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog. Neurobiol 39, 563–607 (1992). - PubMed
Methods-only References
-
- Lipska BK et al. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol. Psychiatry 60, 650–658 (2006). - PubMed
-
- Martin Morgan VO SummarizedExperiment. Bioconductor (2017). doi:10.18129/b9.bioc.summarizedexperiment - DOI
-
- Lun Aaron [Aut C, Davide Risso. SingleCellExperiment. Bioconductor (2017). doi:10.18129/b9.bioc.singlecellexperiment - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
