

Acute BAF perturbation causes immediate changes in chromatin accessibility
- PMID: 33558760
- PMCID: PMC7614082
- DOI: 10.1038/s41588-021-00777-3
Acute BAF perturbation causes immediate changes in chromatin accessibility
Abstract
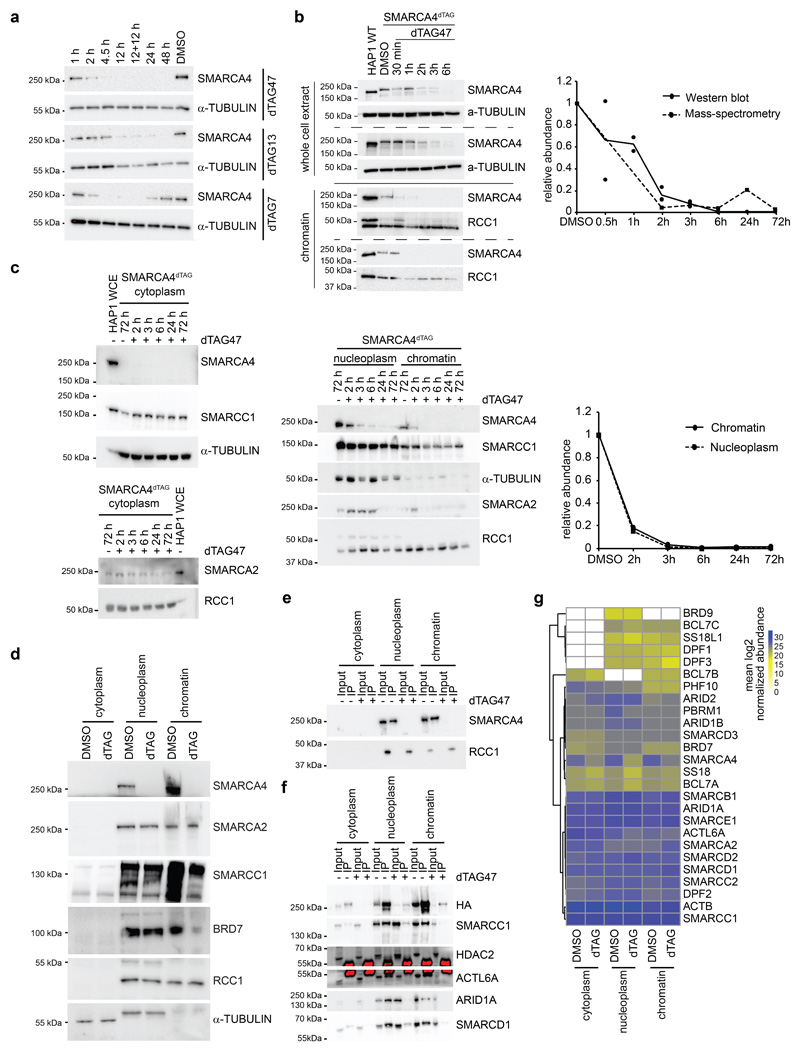
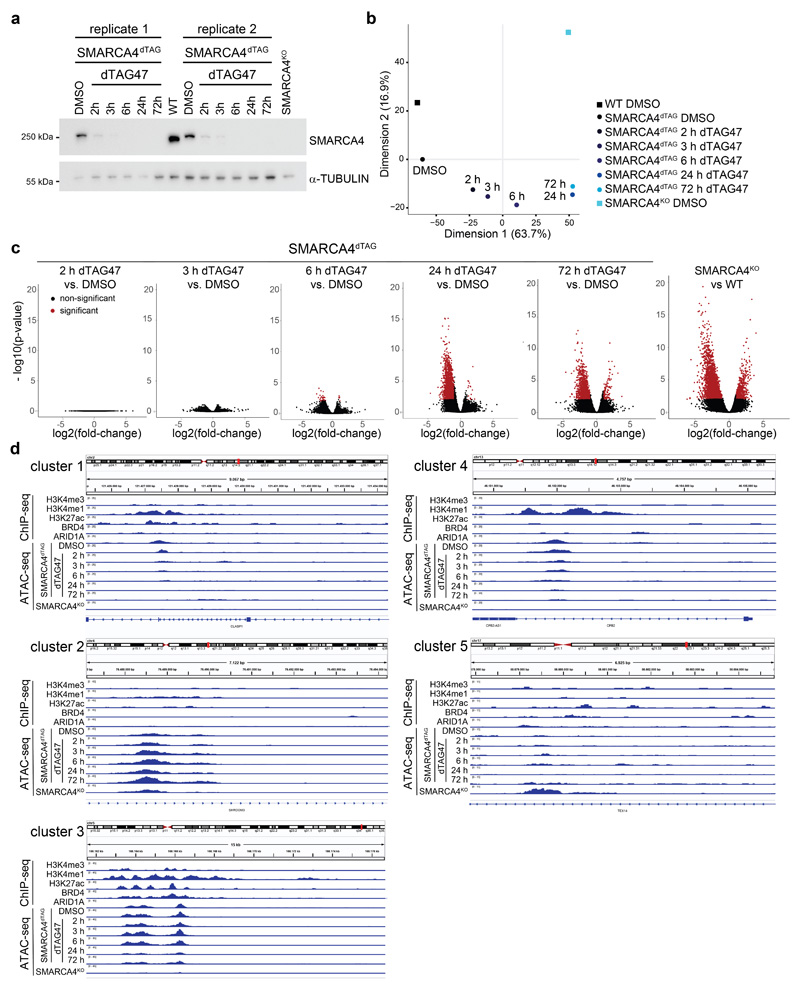
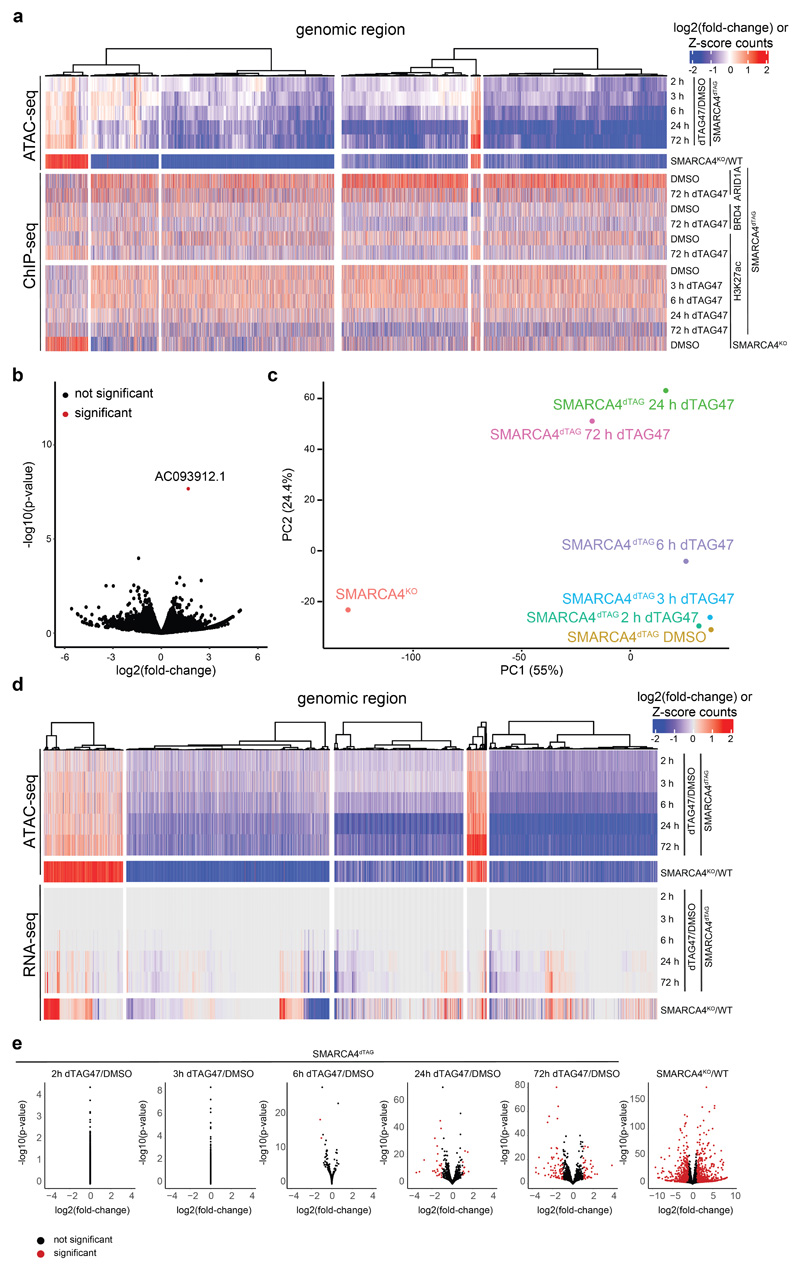
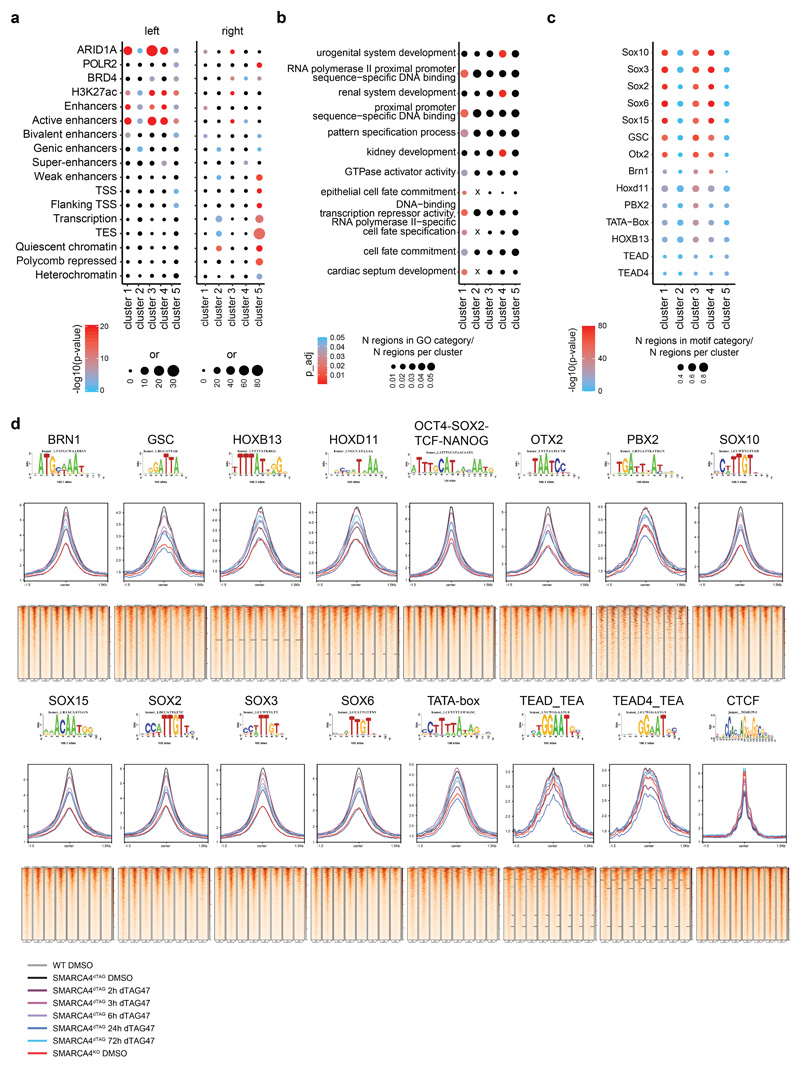
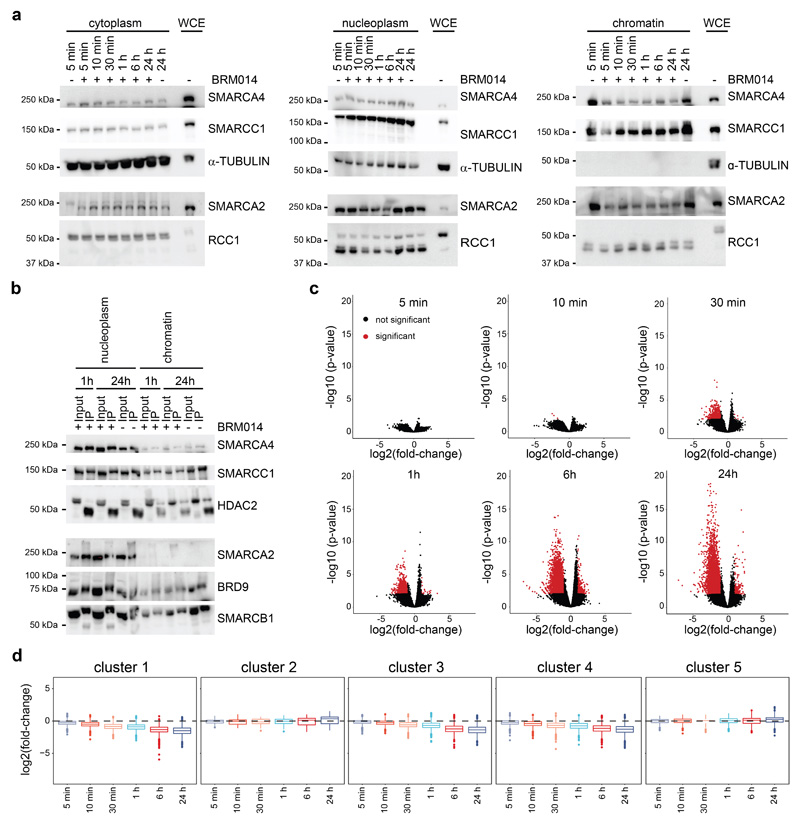
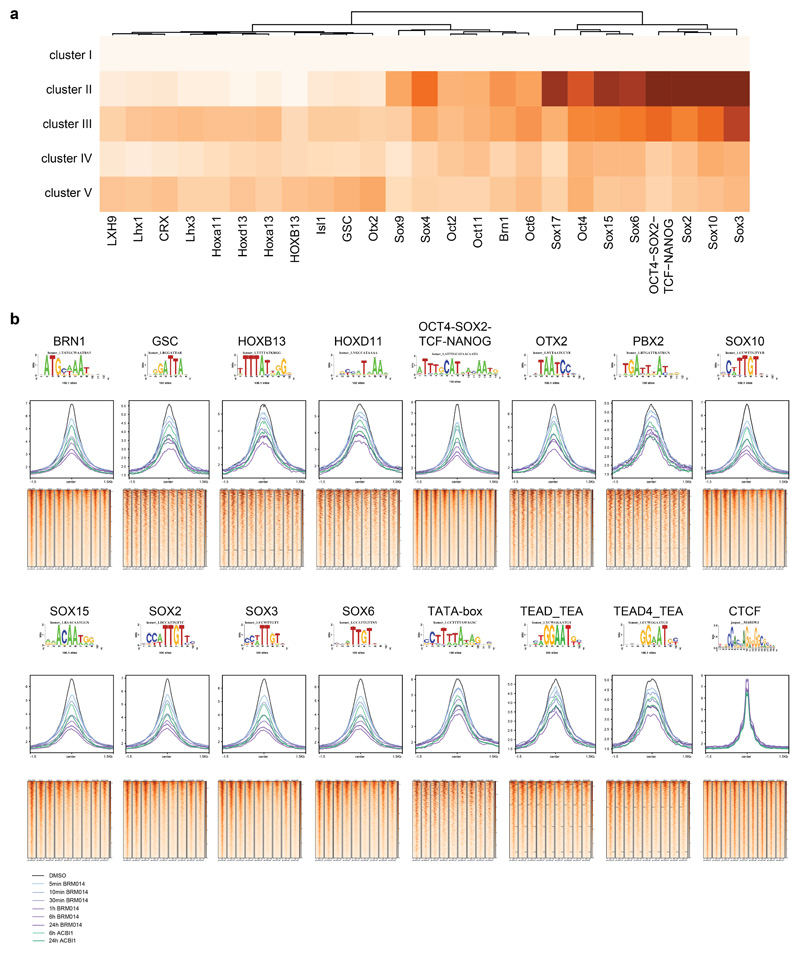
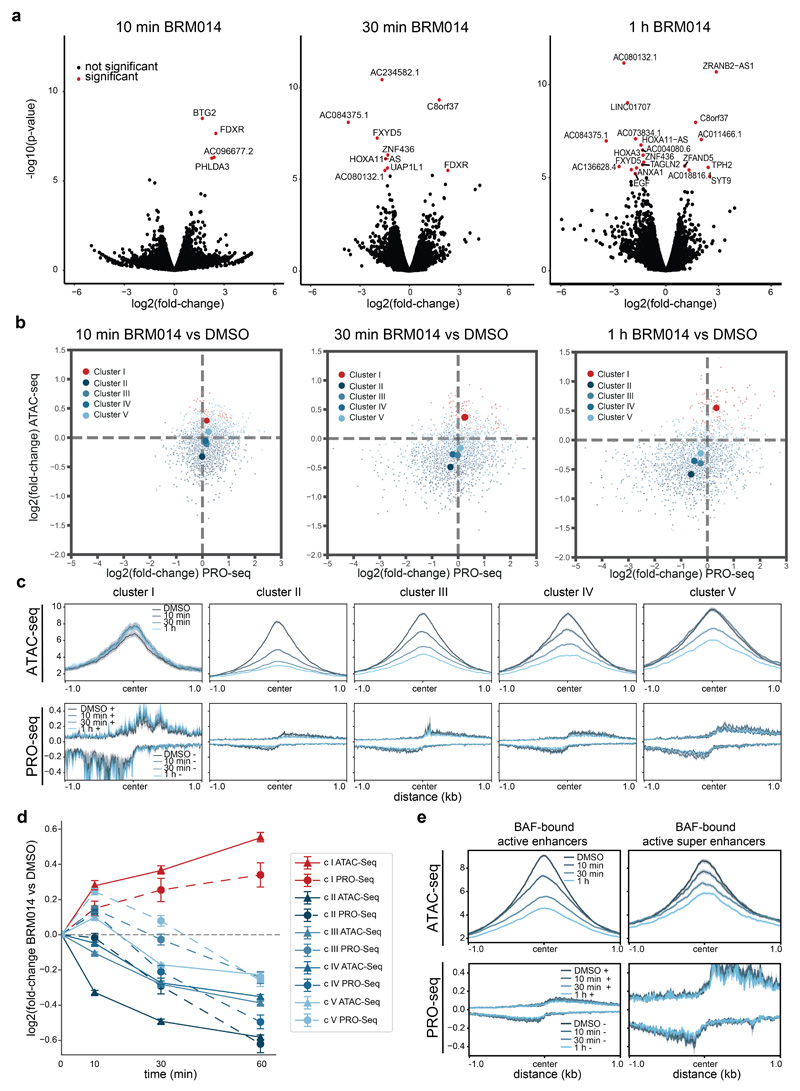
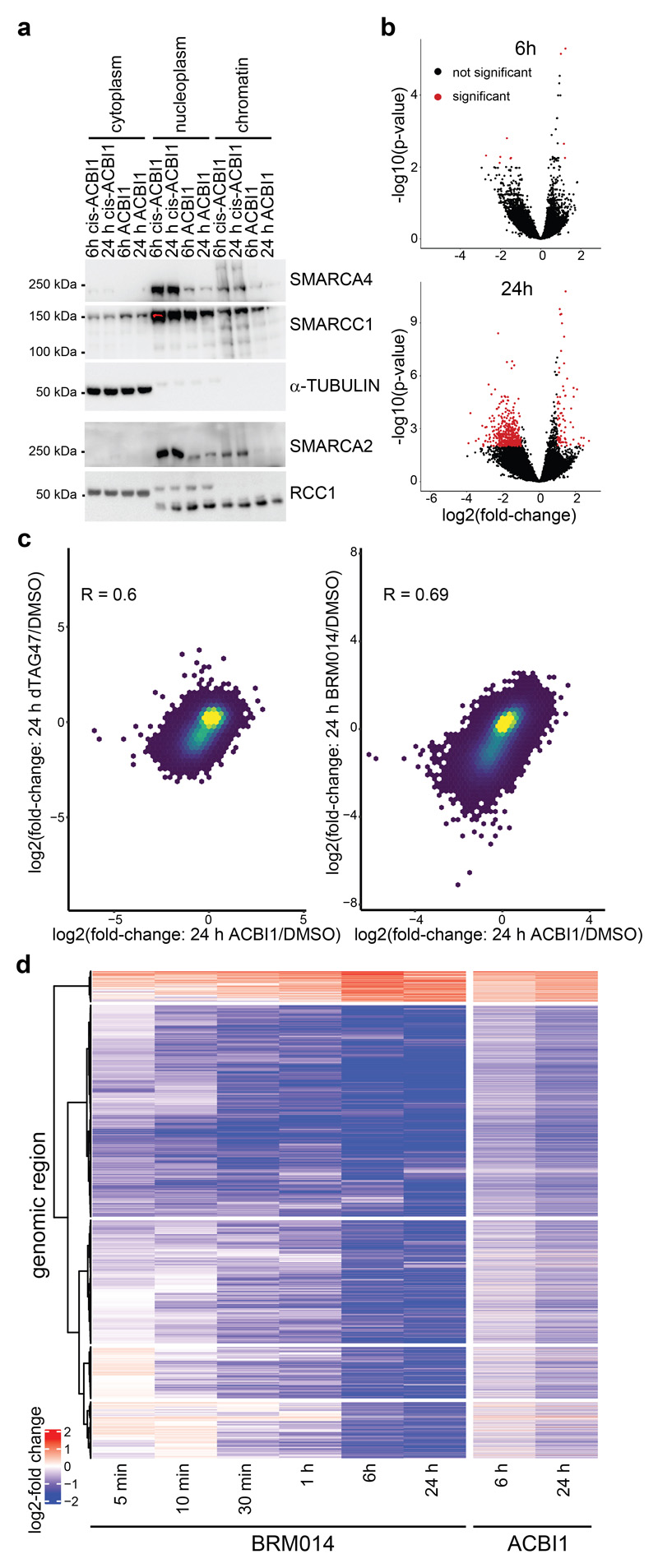
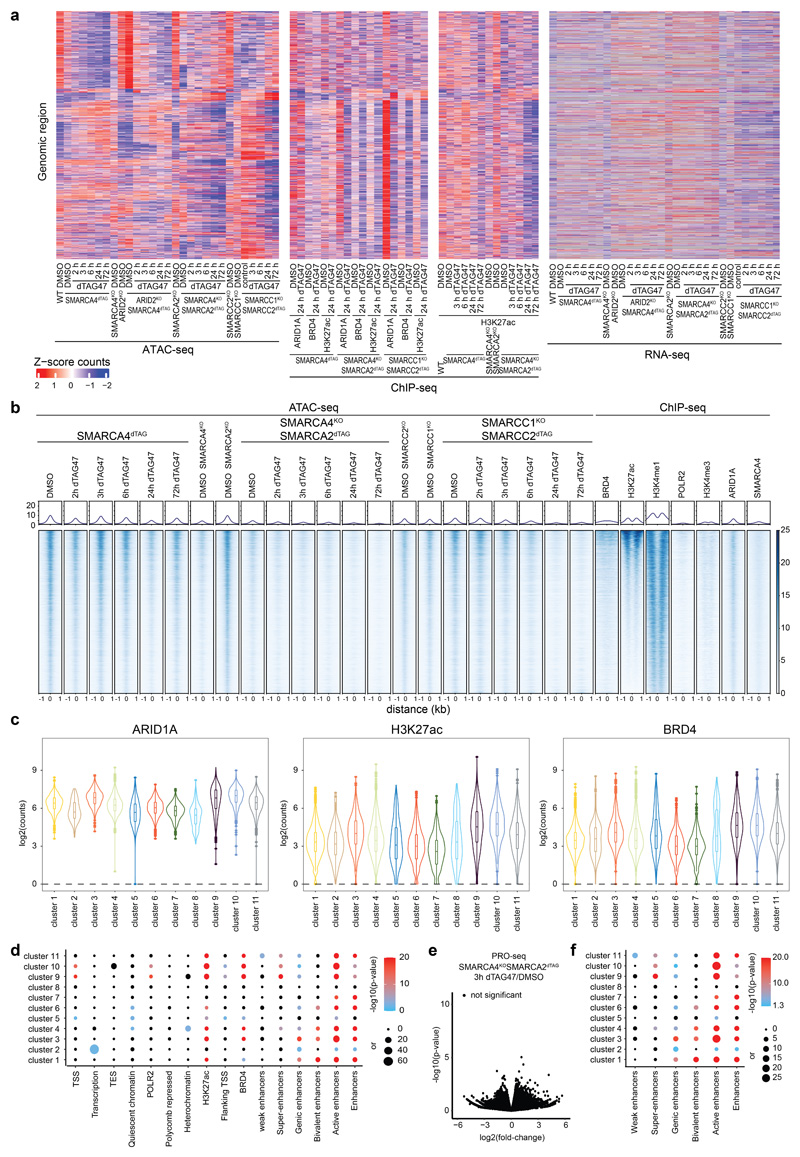
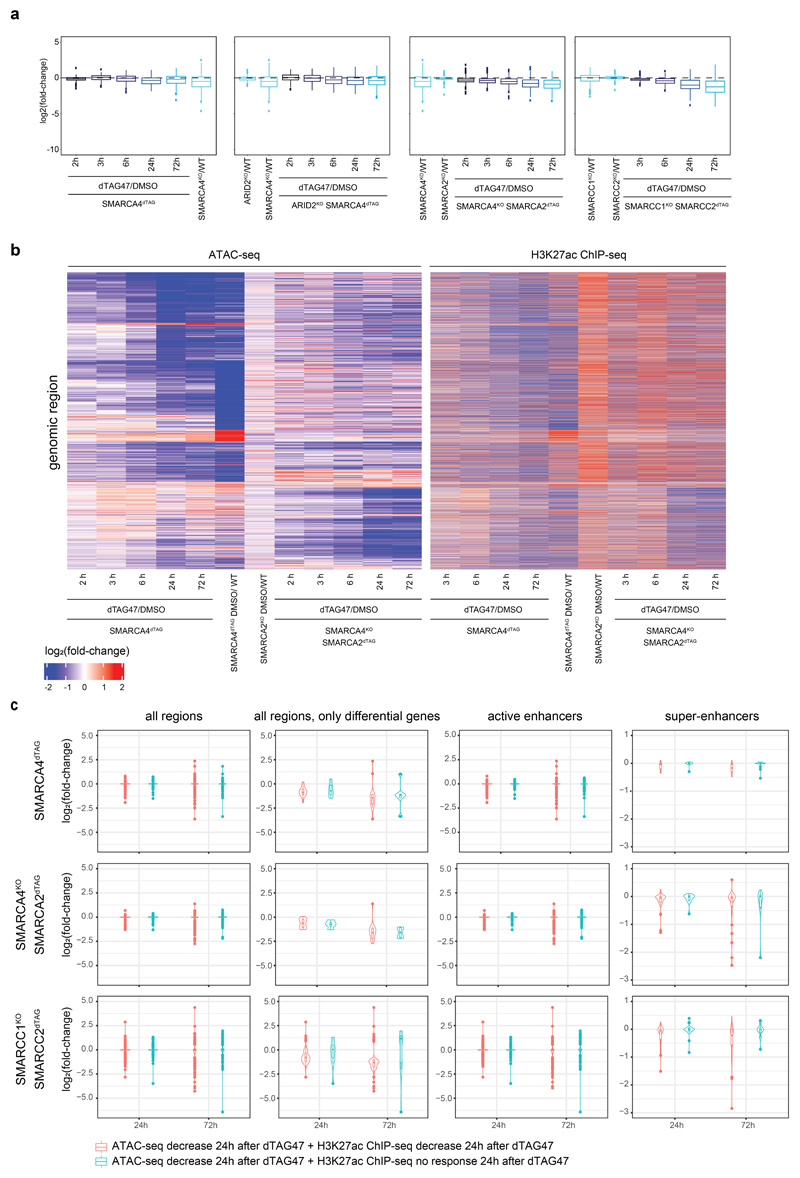
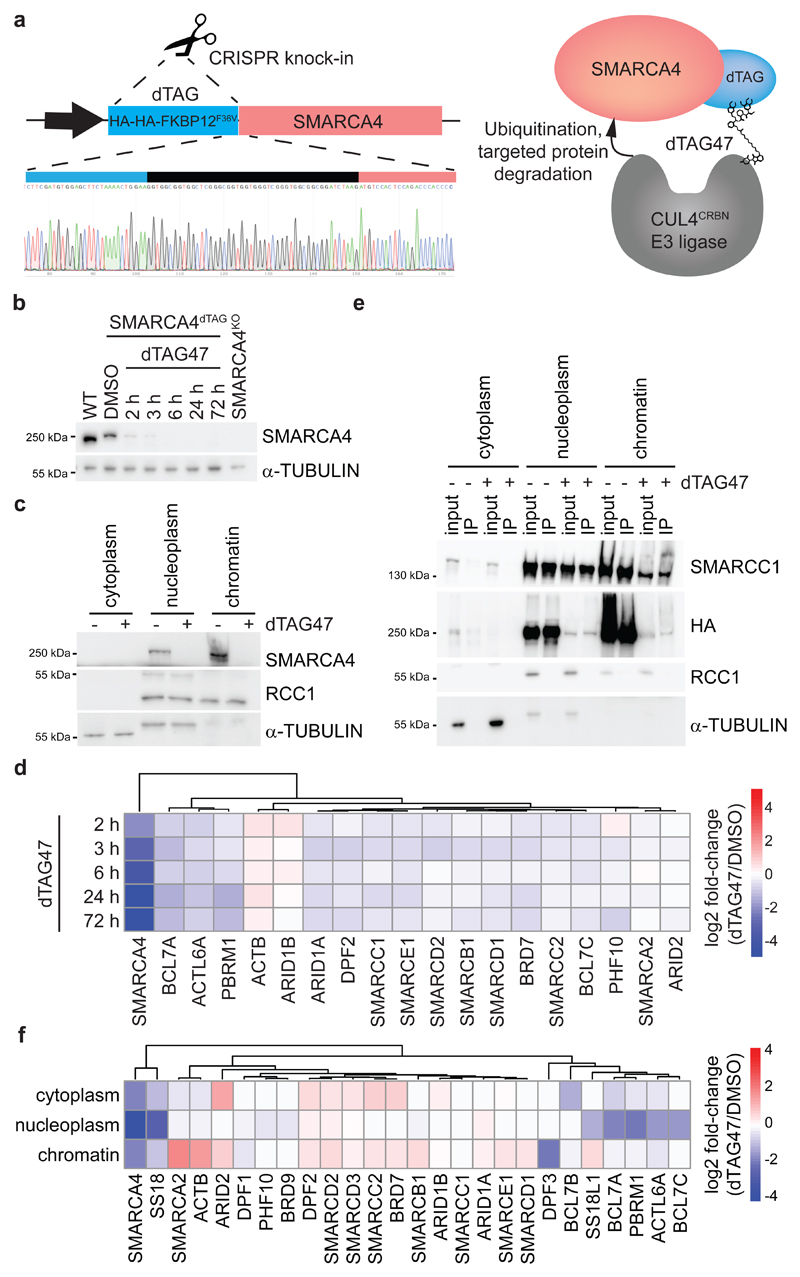
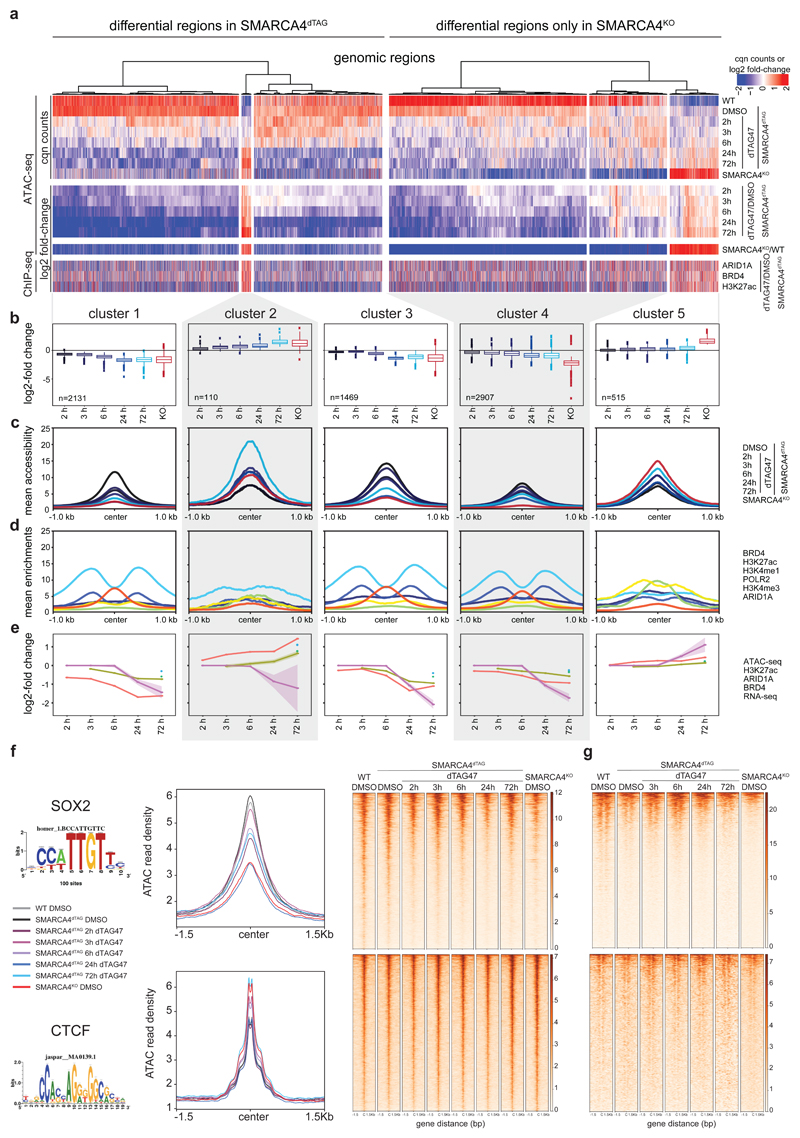
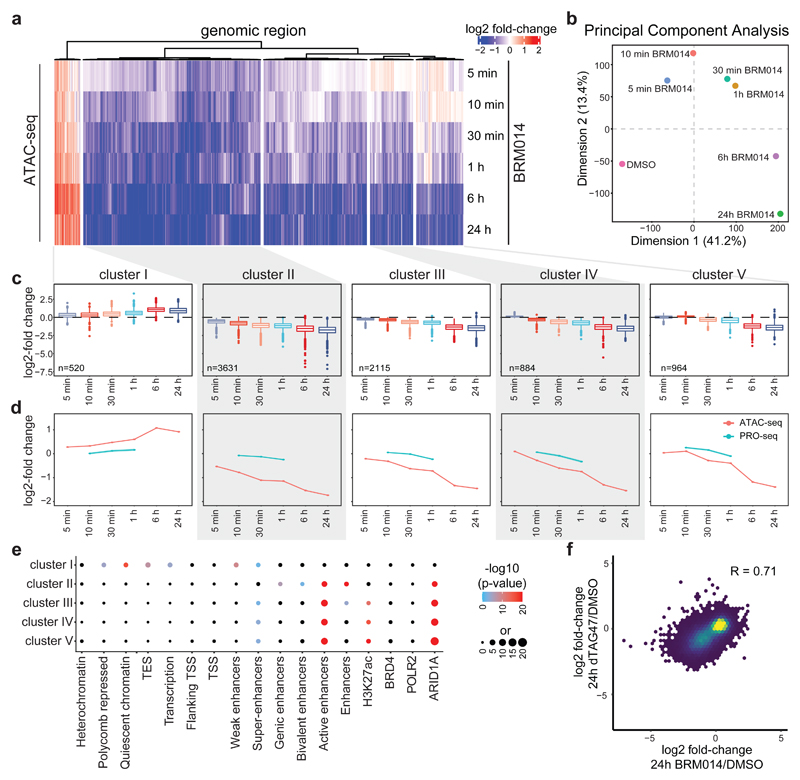
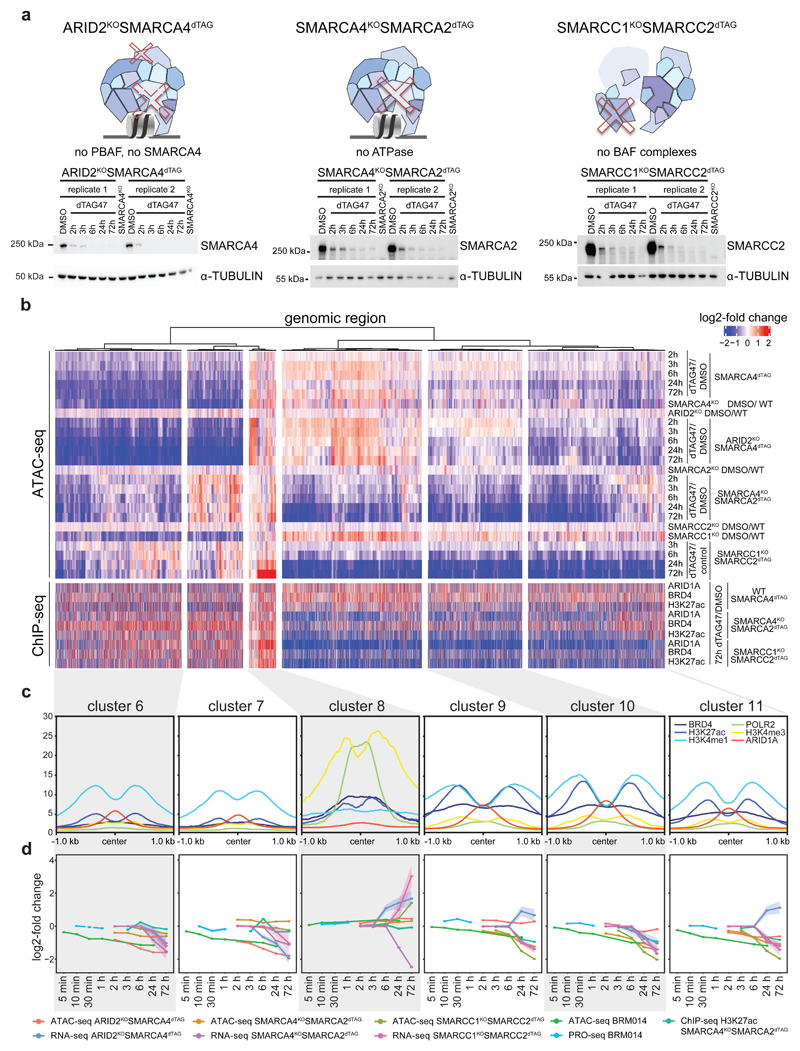
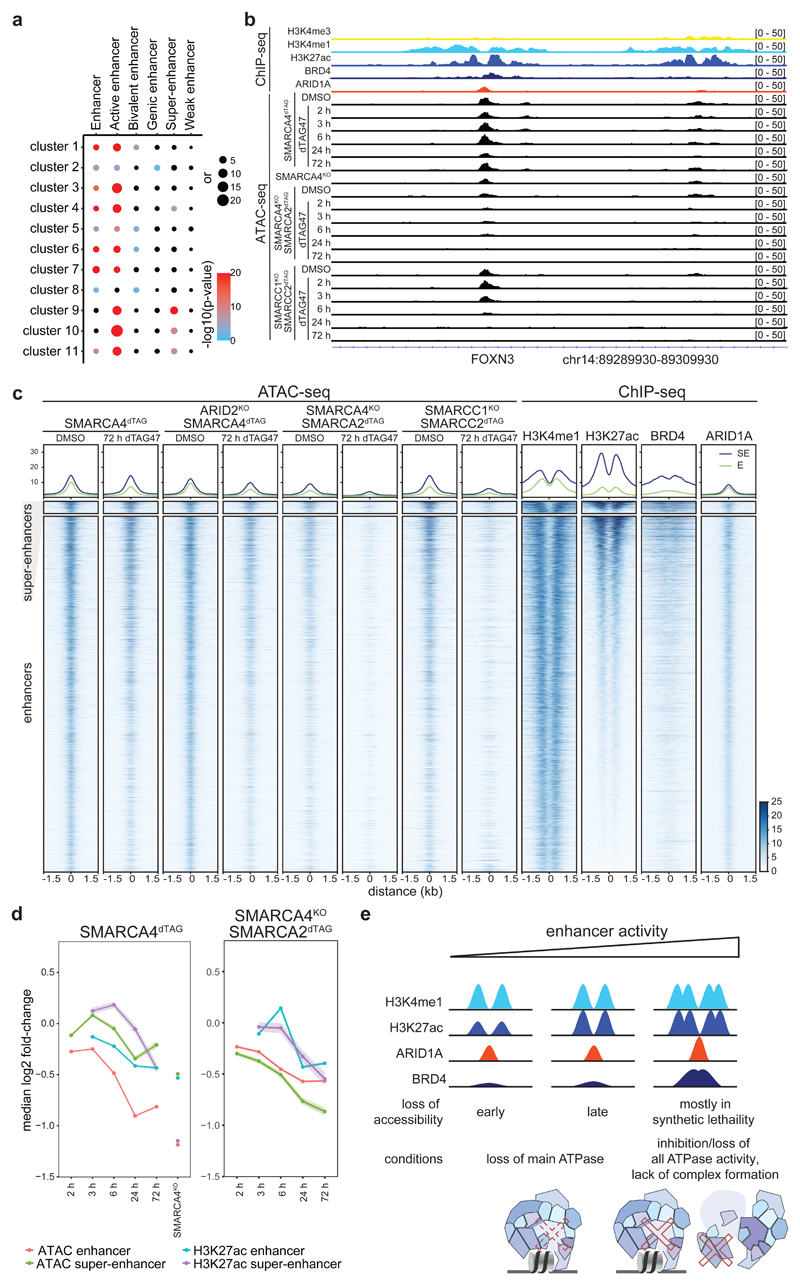
Cancer-associated, loss-of-function mutations in genes encoding subunits of the BRG1/BRM-associated factor (BAF) chromatin-remodeling complexes1-8 often cause drastic chromatin accessibility changes, especially in important regulatory regions9-19. However, it remains unknown how these changes are established over time (for example, immediate consequences or long-term adaptations), and whether they are causative for intracomplex synthetic lethalities, abrogating the formation or activity of BAF complexes9,20-24. In the present study, we use the dTAG system to induce acute degradation of BAF subunits and show that chromatin alterations are established faster than the duration of one cell cycle. Using a pharmacological inhibitor and a chemical degrader of the BAF complex ATPase subunits25,26, we show that maintaining genome accessibility requires constant ATP-dependent remodeling. Completely abolishing BAF complex function by acute degradation of a synthetic lethal subunit in a paralog-deficient background results in an almost complete loss of chromatin accessibility at BAF-controlled sites, especially also at superenhancers, providing a mechanism for intracomplex synthetic lethalities.
Conflict of interest statement
M.P. is an employee of Boehringer Ingelheim RCV GmbH & Co KG. G.W. and S.K. are co-founders and shareholders of Proxygen GmbH.
Figures
Comment in
-
Chromatin openness requires continuous SWI/SNF activity.Nat Genet. 2021 Mar;53(3):263-264. doi: 10.1038/s41588-021-00781-7. Nat Genet. 2021. PMID: 33558759 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
