

Tracing the origins of SARS-COV-2 in coronavirus phylogenies: a review
- PMID: 33558807
- PMCID: PMC7859469
- DOI: 10.1007/s10311-020-01151-1
Tracing the origins of SARS-COV-2 in coronavirus phylogenies: a review
Abstract
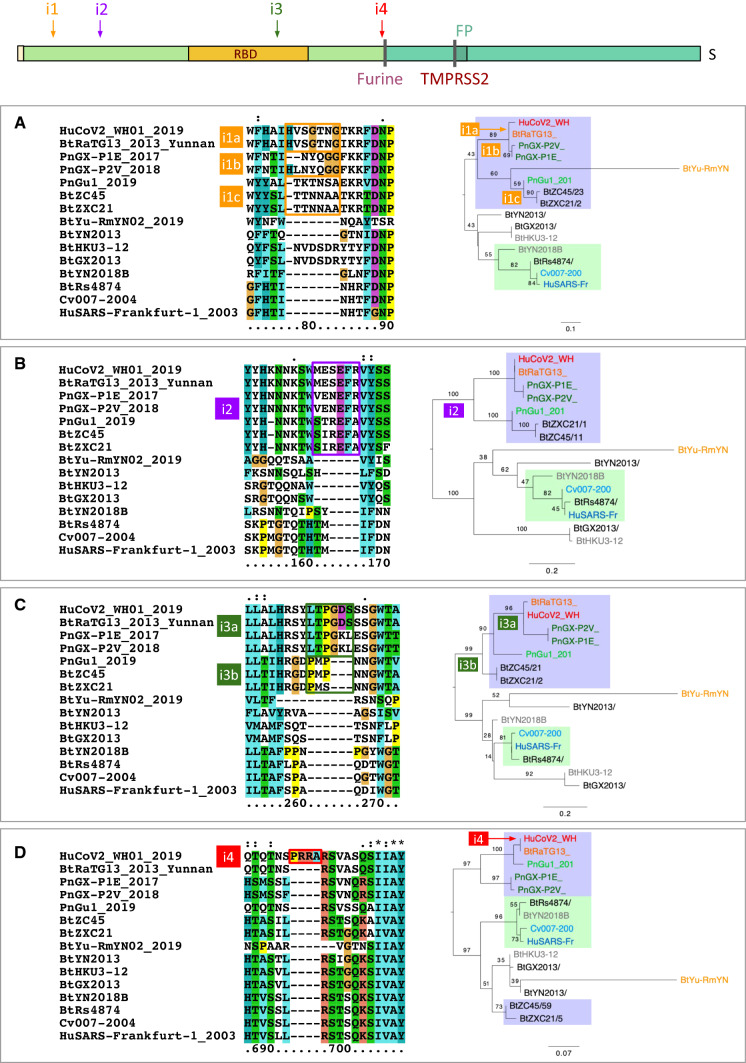
SARS-CoV-2 is a new human coronavirus (CoV), which emerged in China in late 2019 and is responsible for the global COVID-19 pandemic that caused more than 97 million infections and 2 million deaths in 12 months. Understanding the origin of this virus is an important issue, and it is necessary to determine the mechanisms of viral dissemination in order to contain future epidemics. Based on phylogenetic inferences, sequence analysis and structure-function relationships of coronavirus proteins, informed by the knowledge currently available on the virus, we discuss the different scenarios on the origin-natural or synthetic-of the virus. The data currently available are not sufficient to firmly assert whether SARS-CoV2 results from a zoonotic emergence or from an accidental escape of a laboratory strain. This question needs to be solved because it has important consequences on the risk/benefit balance of our interactions with ecosystems, on intensive breeding of wild and domestic animals, on some laboratory practices and on scientific policy and biosafety regulations. Regardless of COVID-19 origin, studying the evolution of the molecular mechanisms involved in the emergence of pandemic viruses is essential to develop therapeutic and vaccine strategies and to prevent future zoonoses. This article is a translation and update of a French article published in Médecine/Sciences, August/September 2020 (10.1051/medsci/2020123).
Supplementary information: The online version of this article (10.1007/s10311-020-01151-1) contains supplementary material, which is available to authorized users.
Keywords: Bioinformatics; Biosafety; Coronavirus; Covid-19; Furin; Gain of function; Genome analysis; Pandemic; Phylogeny; SARS-CoV-2; Spike protein; Virology; Zoonosis.
© The Author(s) 2021.
Figures
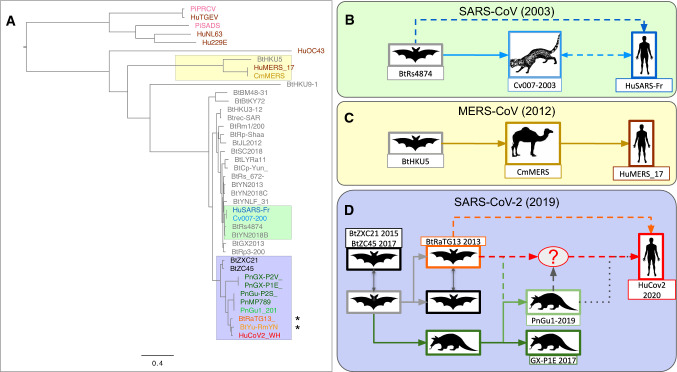
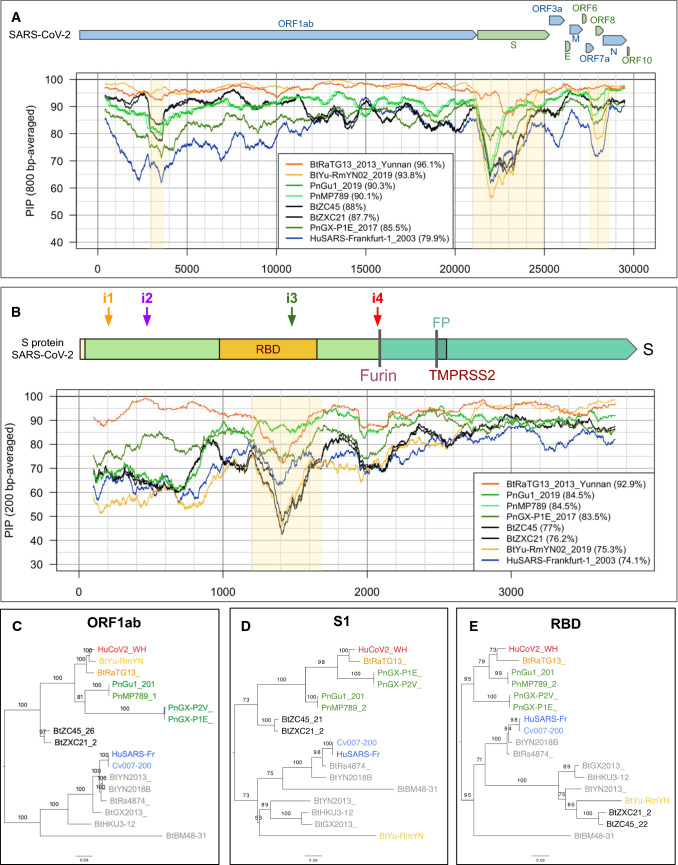
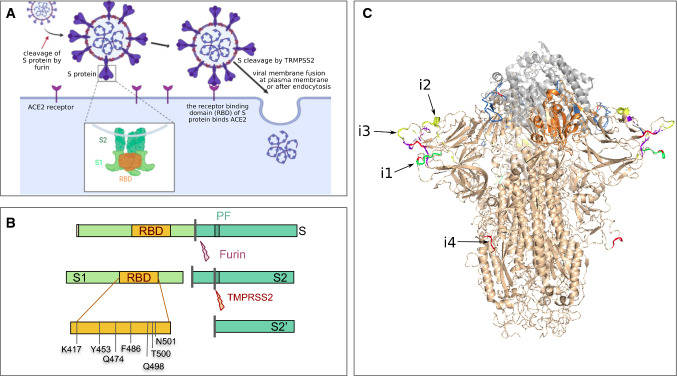
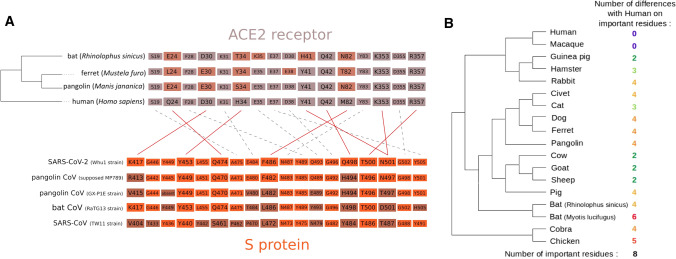

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous