

Proteomic investigation reveals dominant alterations of neutrophil degranulation and mRNA translation pathways in patients with COVID-19
- PMID: 33558857
- PMCID: PMC7857979
- DOI: 10.1016/j.isci.2021.102135
Proteomic investigation reveals dominant alterations of neutrophil degranulation and mRNA translation pathways in patients with COVID-19
Abstract
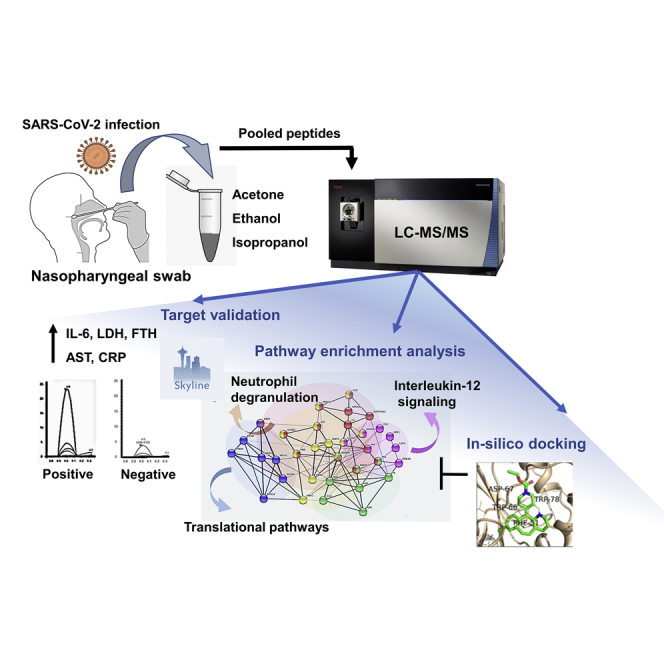
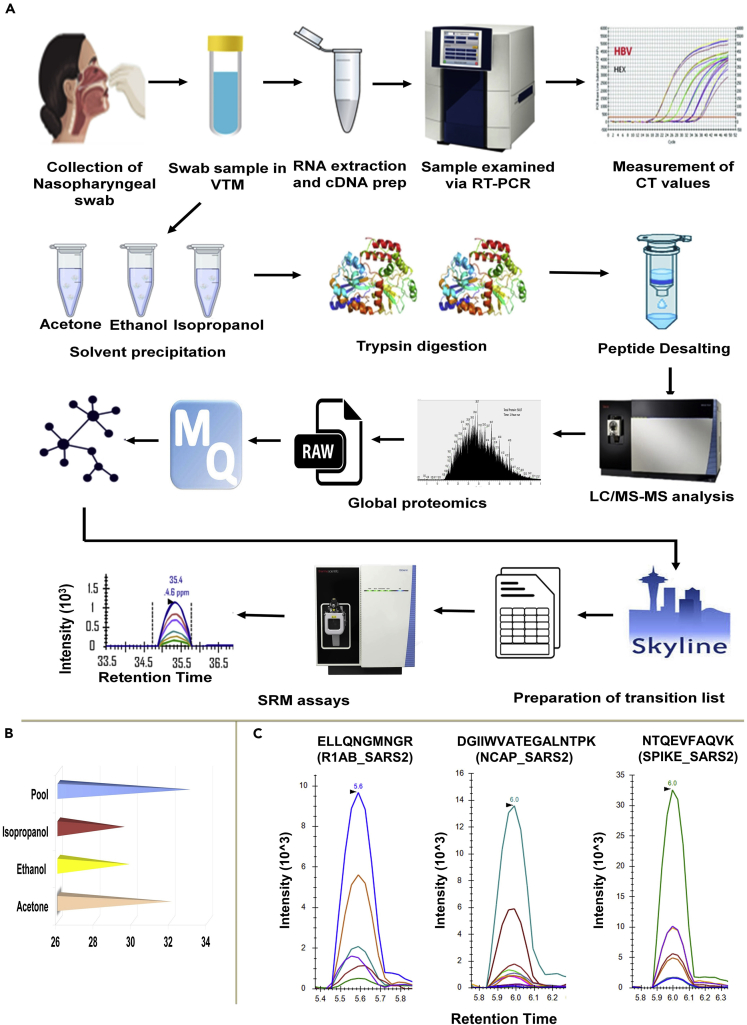
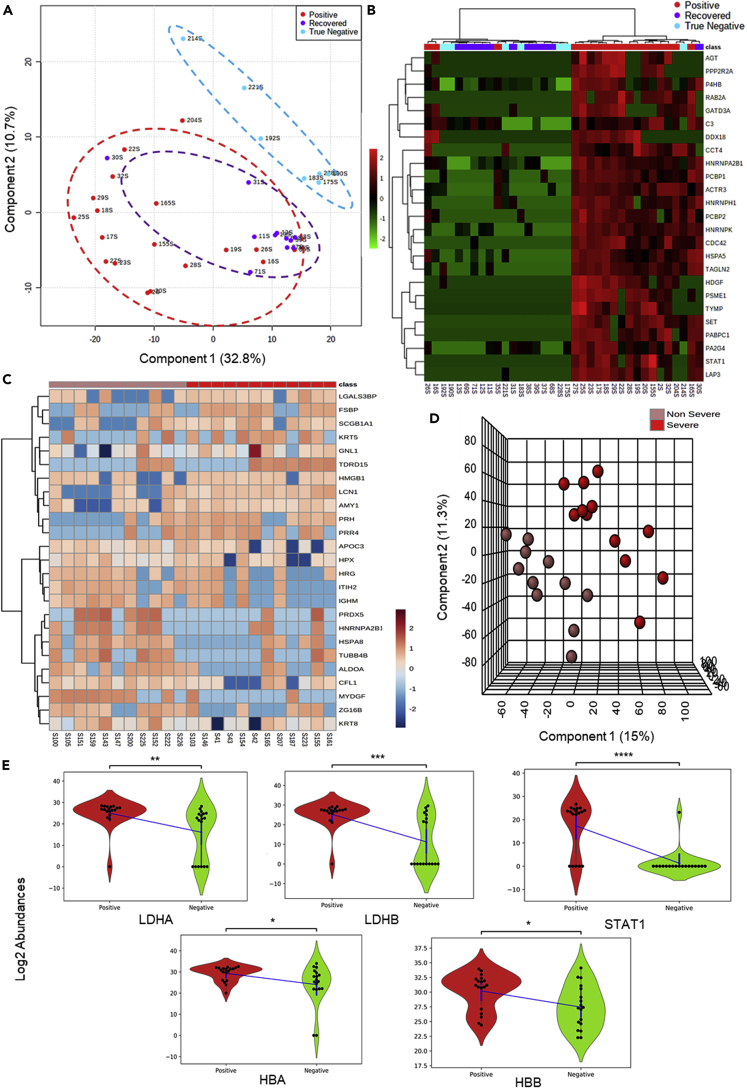
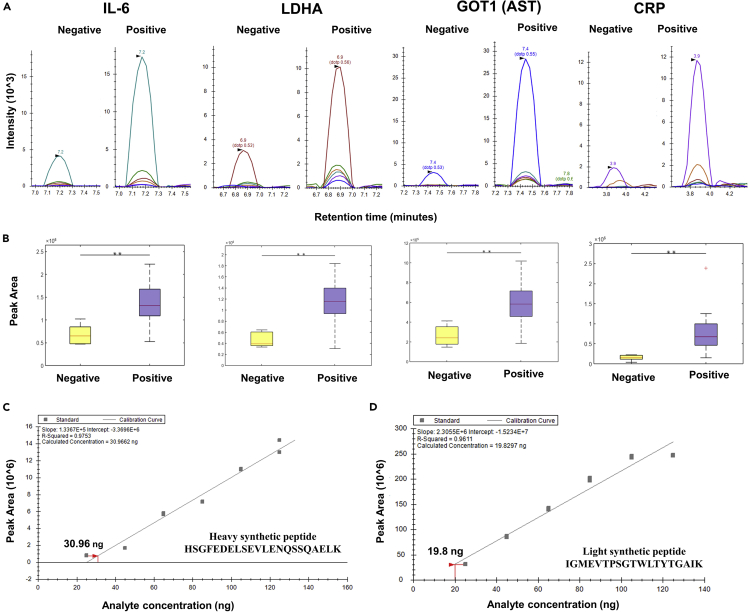
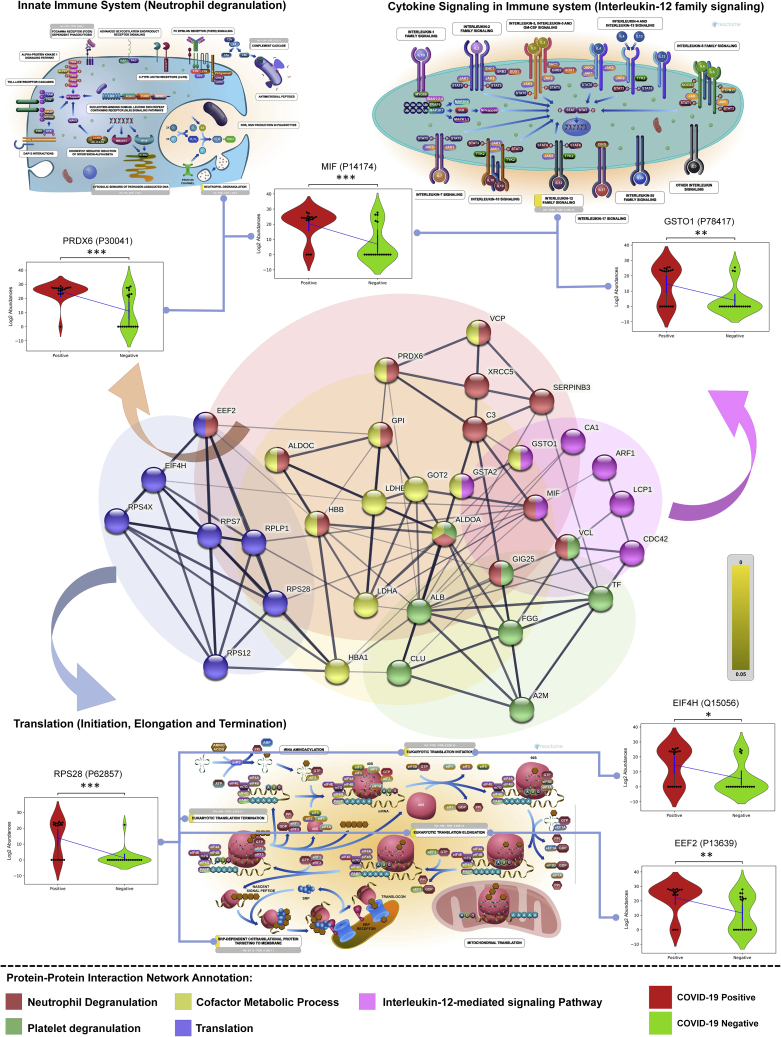
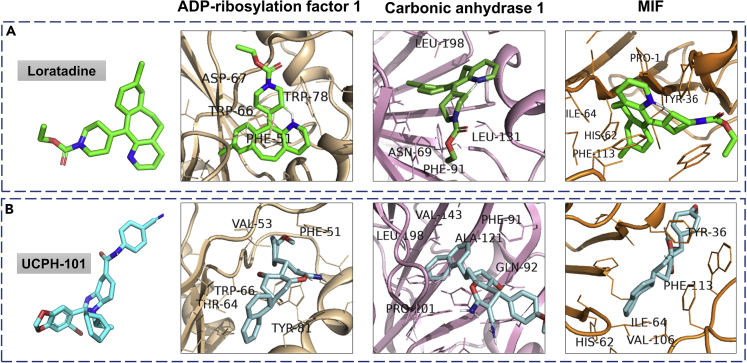
The altered molecular proteins and pathways in response to COVID-19 infection are still unclear. Here, we performed a comprehensive proteomics-based investigation of nasopharyngeal swab samples from patients with COVID-19 to study the host response by employing simple extraction strategies. Few of the host proteins such as interleukin-6, L-lactate dehydrogenase, C-reactive protein, Ferritin, and aspartate aminotransferase were found to be upregulated only in COVID-19-positive patients using targeted multiple reaction monitoring studies. The most important pathways identified by enrichment analysis were neutrophil degranulation, interleukin-12 signaling pathways, and mRNA translation of proteins thus providing the detailed investigation of host response in COVID-19 infection. Thus, we conclude that mass spectrometry-detected host proteins have a potential for disease severity progression; however, suitable validation strategies should be deployed for the clinical translation. Furthermore, the in silico docking of potential drugs with host proteins involved in the interleukin-12 signaling pathway might aid in COVID-19 therapeutic interventions.
Keywords: molecular biology; proteomics; specimen.
© 2021 The Author(s).
Conflict of interest statement
The authors have filed an Indian patent related to this work “Protein markers and method for prognosis of COVID-19 in individuals”. (Application number: 202021034688). The authors declare no competing interests. “Proteomics-based method for detection of Coronavirus in a sample” (Application number 202021034687).
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
