

An ancestral 10-bp repeat expansion in VWA1 causes recessive hereditary motor neuropathy
- PMID: 33559681
- PMCID: PMC8263055
- DOI: 10.1093/brain/awaa420
An ancestral 10-bp repeat expansion in VWA1 causes recessive hereditary motor neuropathy
Abstract
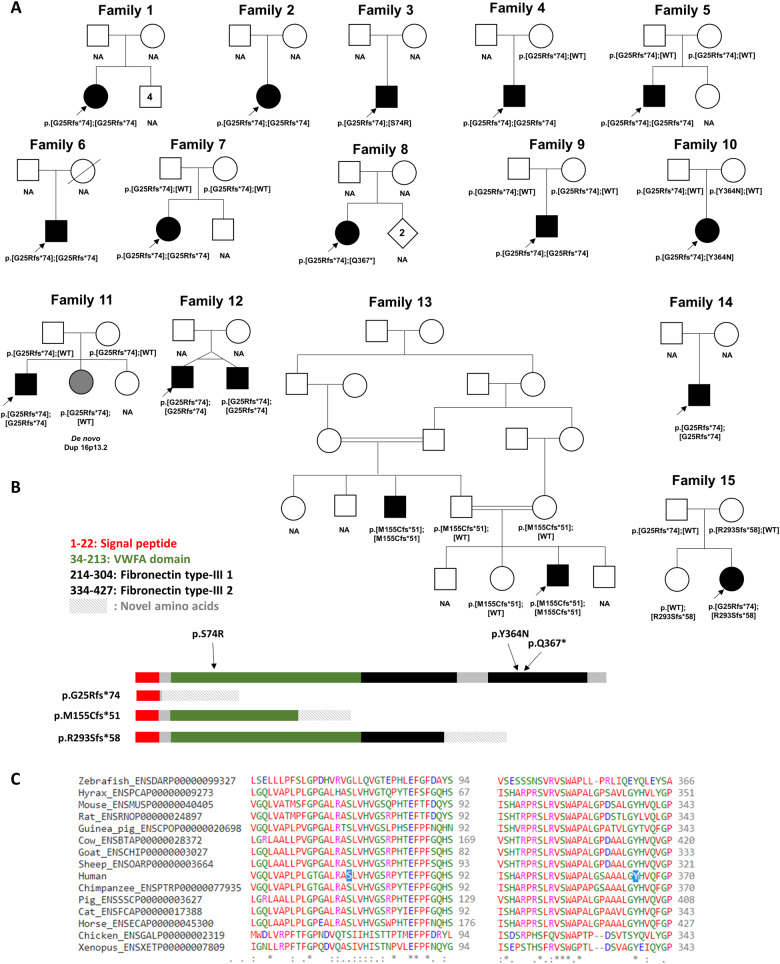
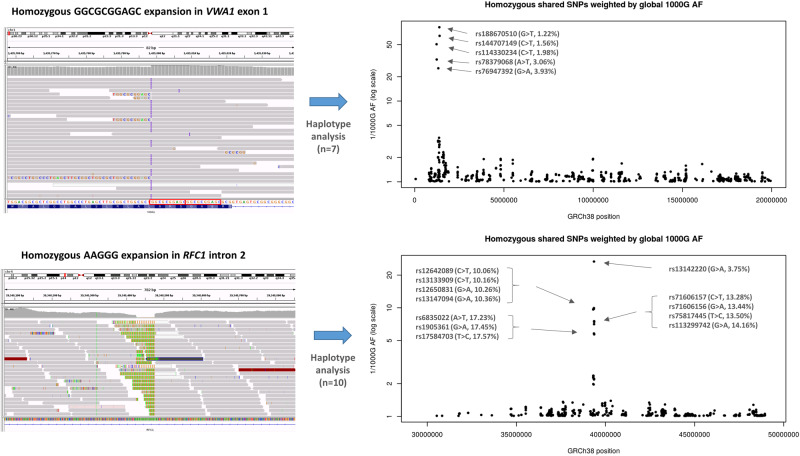
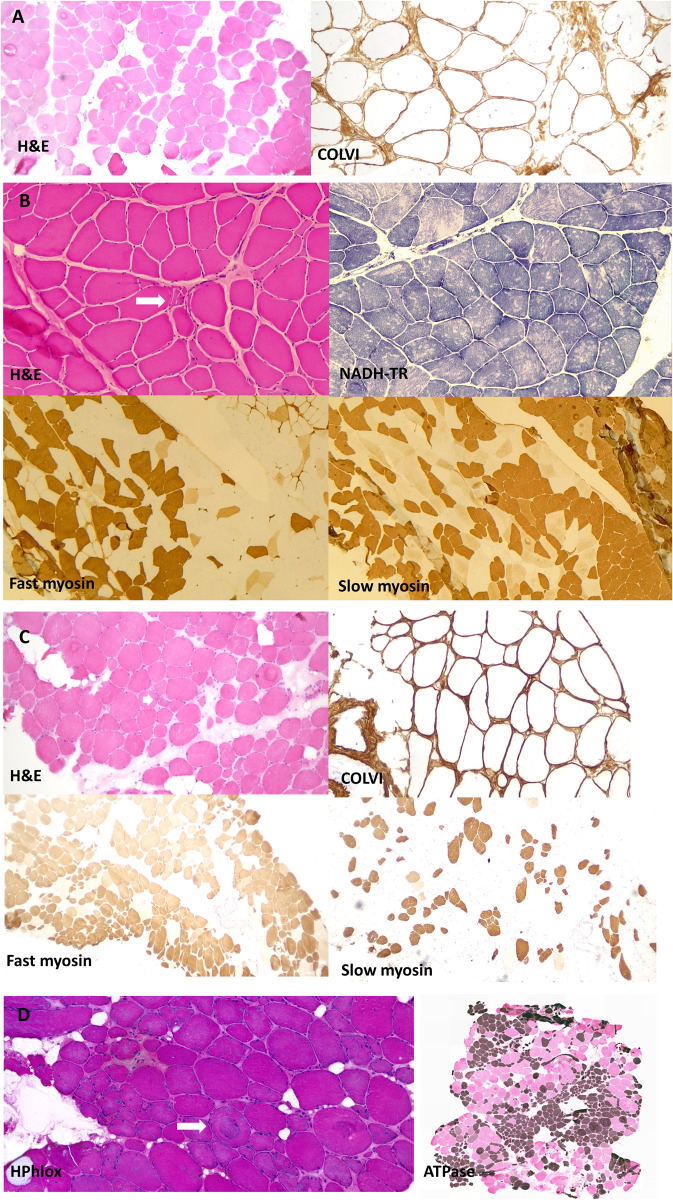
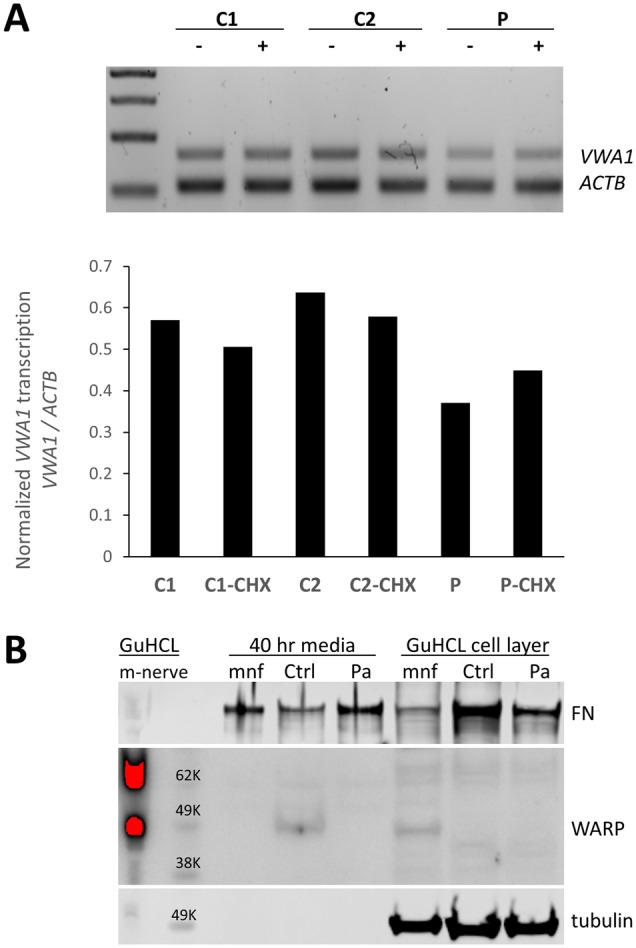
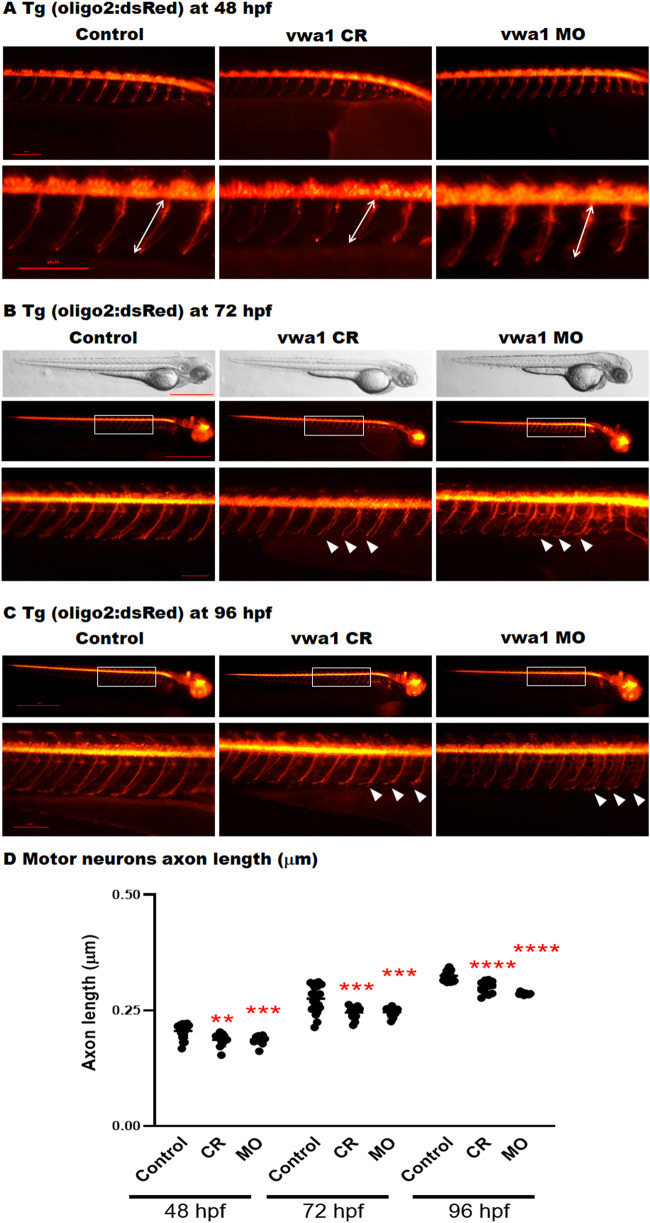
The extracellular matrix comprises a network of macromolecules such as collagens, proteoglycans and glycoproteins. VWA1 (von Willebrand factor A domain containing 1) encodes a component of the extracellular matrix that interacts with perlecan/collagen VI, appears to be involved in stabilizing extracellular matrix structures, and demonstrates high expression levels in tibial nerve. Vwa1-deficient mice manifest with abnormal peripheral nerve structure/function; however, VWA1 variants have not previously been associated with human disease. By interrogating the genome sequences of 74 180 individuals from the 100K Genomes Project in combination with international gene-matching efforts and targeted sequencing, we identified 17 individuals from 15 families with an autosomal-recessive, non-length dependent, hereditary motor neuropathy and rare biallelic variants in VWA1. A single disease-associated allele p.(G25Rfs*74), a 10-bp repeat expansion, was observed in 14/15 families and was homozygous in 10/15. Given an allele frequency in European populations approaching 1/1000, the seven unrelated homozygote individuals ascertained from the 100K Genomes Project represents a substantial enrichment above expected. Haplotype analysis identified a shared 220 kb region suggesting that this founder mutation arose >7000 years ago. A wide age-range of patients (6-83 years) helped delineate the clinical phenotype over time. The commonest disease presentation in the cohort was an early-onset (mean 2.0 ± 1.4 years) non-length-dependent axonal hereditary motor neuropathy, confirmed on electrophysiology, which will have to be differentiated from other predominantly or pure motor neuropathies and neuronopathies. Because of slow disease progression, ambulation was largely preserved. Neurophysiology, muscle histopathology, and muscle MRI findings typically revealed clear neurogenic changes with single isolated cases displaying additional myopathic process. We speculate that a few findings of myopathic changes might be secondary to chronic denervation rather than indicating an additional myopathic disease process. Duplex reverse transcription polymerase chain reaction and immunoblotting using patient fibroblasts revealed that the founder allele results in partial nonsense mediated decay and an absence of detectable protein. CRISPR and morpholino vwa1 modelling in zebrafish demonstrated reductions in motor neuron axonal growth, synaptic formation in the skeletal muscles and locomotive behaviour. In summary, we estimate that biallelic variants in VWA1 may be responsible for up to 1% of unexplained hereditary motor neuropathy cases in Europeans. The detailed clinical characterization provided here will facilitate targeted testing on suitable patient cohorts. This novel disease gene may have previously evaded detection because of high GC content, consequential low coverage and computational difficulties associated with robustly detecting repeat-expansions. Reviewing previously unsolved exomes using lower QC filters may generate further diagnoses.
Keywords: EMG; genetics: neuropathy; hereditary motor and sensory neuropathies; nerve conduction studies; whole-genome sequencing.
© The Author(s) (2021). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
Genetic alterations of VWA1: a new link between extracellular matrix and neuromuscular diseases.Brain. 2021 Mar 3;144(2):362-365. doi: 10.1093/brain/awaa464. Brain. 2021. PMID: 33693694 No abstract available.
References
-
- Allen JM, Brachvogel B, Farlie PG, Fitzgerald J, Bateman JF.. The extracellular matrix protein WARP is a novel component of a distinct subset of basement membranes. Matrix Biol 2008; 27: 295–305. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
