

Visualizing population structure with variational autoencoders
- PMID: 33561250
- PMCID: PMC8022710
- DOI: 10.1093/g3journal/jkaa036
Visualizing population structure with variational autoencoders
Abstract
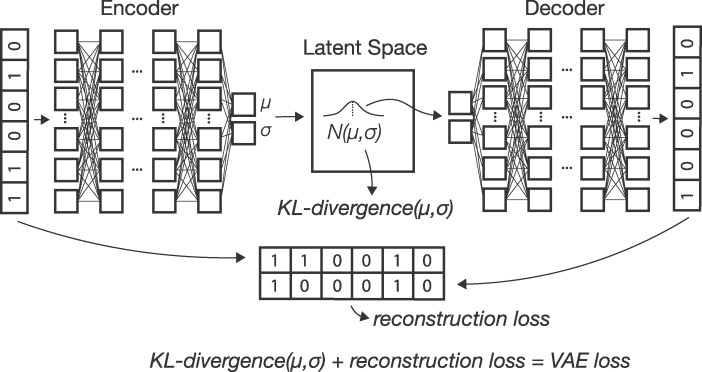
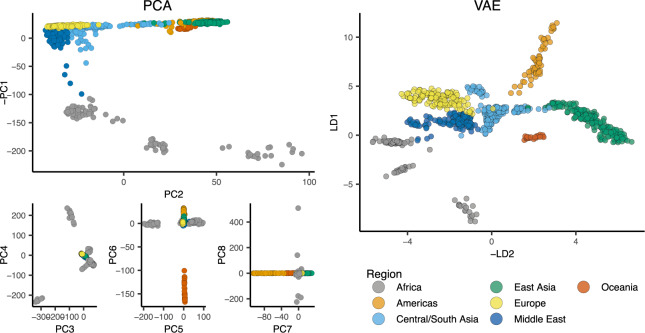
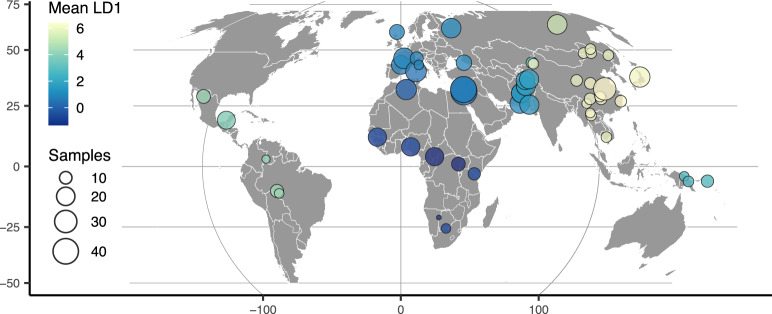
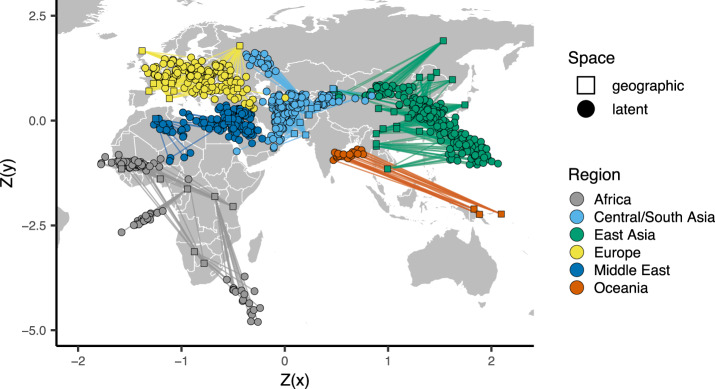
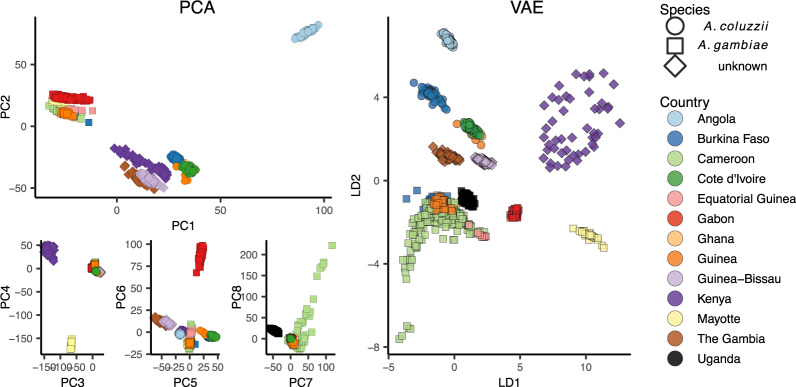
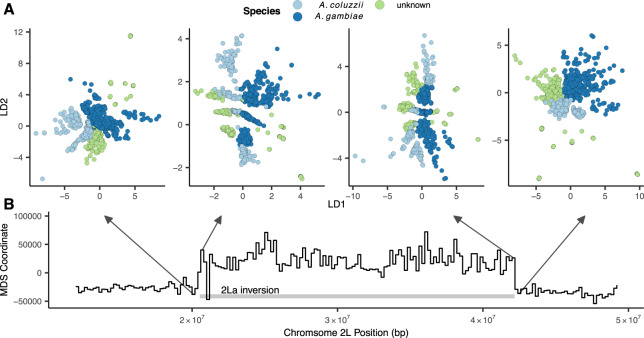
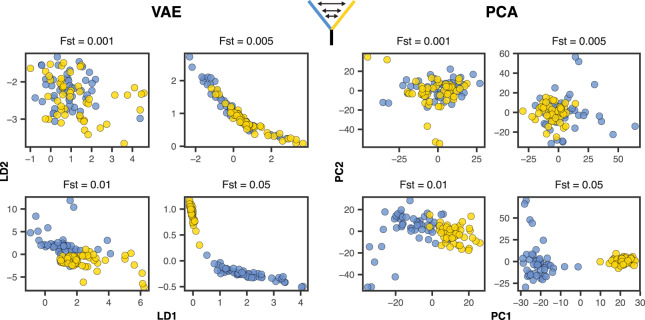
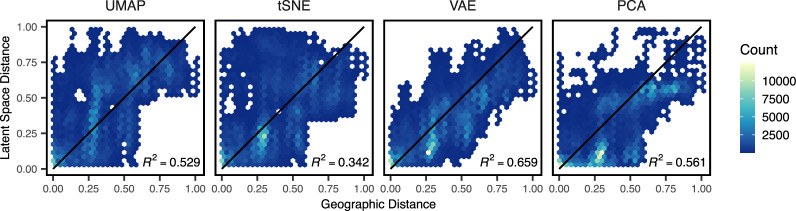
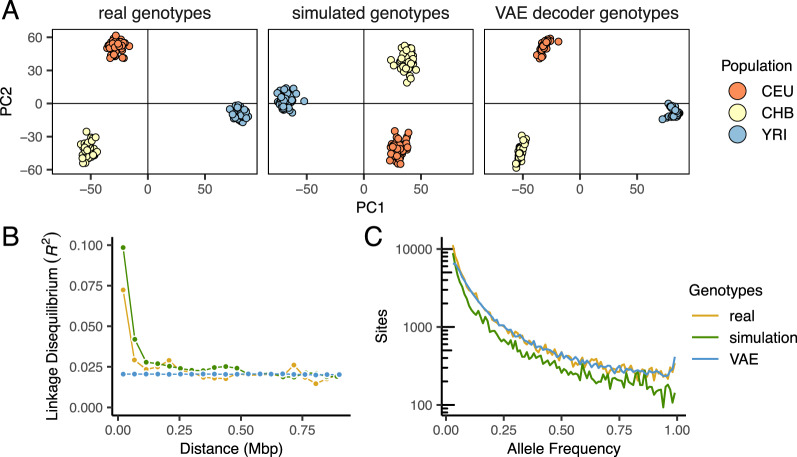
Dimensionality reduction is a common tool for visualization and inference of population structure from genotypes, but popular methods either return too many dimensions for easy plotting (PCA) or fail to preserve global geometry (t-SNE and UMAP). Here we explore the utility of variational autoencoders (VAEs)-generative machine learning models in which a pair of neural networks seek to first compress and then recreate the input data-for visualizing population genetic variation. VAEs incorporate nonlinear relationships, allow users to define the dimensionality of the latent space, and in our tests preserve global geometry better than t-SNE and UMAP. Our implementation, which we call popvae, is available as a command-line python program at github.com/kr-colab/popvae. The approach yields latent embeddings that capture subtle aspects of population structure in humans and Anopheles mosquitoes, and can generate artificial genotypes characteristic of a given sample or population.
Keywords: data visualization; deep learning; machine learning; neural network; pca; population genetics; population structure; variational autoencoder.
© The Author(s) 2021. Published by Oxford University Press on behalf of Genetics Society of America.
Figures
References
-
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, et al.2015. TensorFlow: large-scale machine learning on heterogeneous systems. Software available from tensorflow.org. (Accessed: 2020 October)
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous