

Mutations in the RAS/MAPK Pathway Drive Replication Repair-Deficient Hypermutated Tumors and Confer Sensitivity to MEK Inhibition
- PMID: 33563663
- PMCID: PMC8406556
- DOI: 10.1158/2159-8290.CD-20-1050
Mutations in the RAS/MAPK Pathway Drive Replication Repair-Deficient Hypermutated Tumors and Confer Sensitivity to MEK Inhibition
Abstract
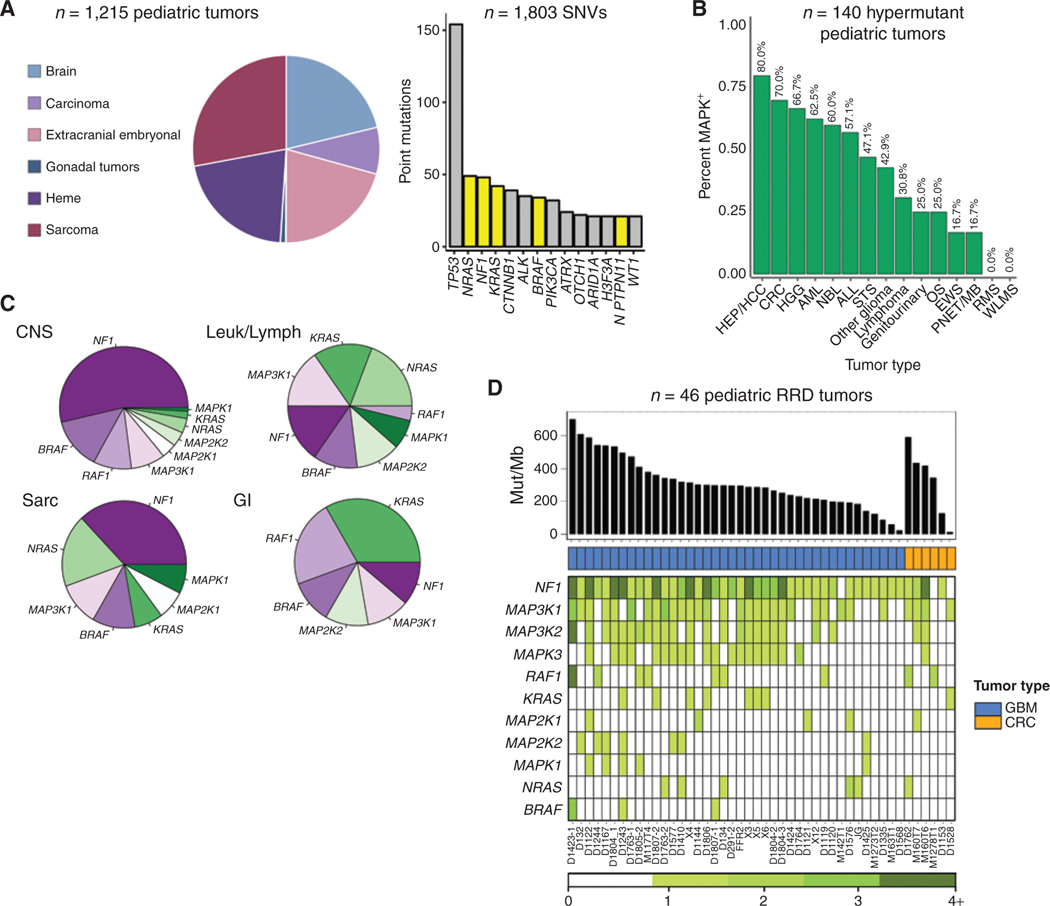
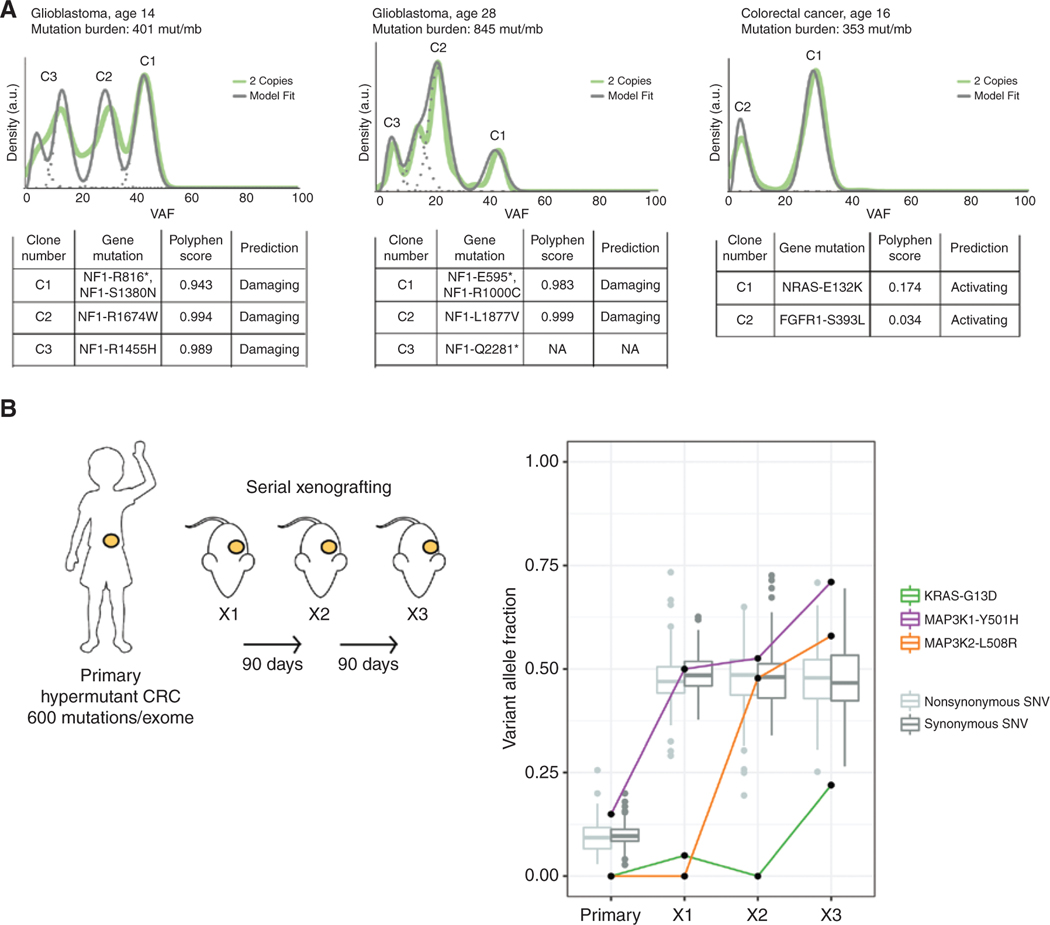
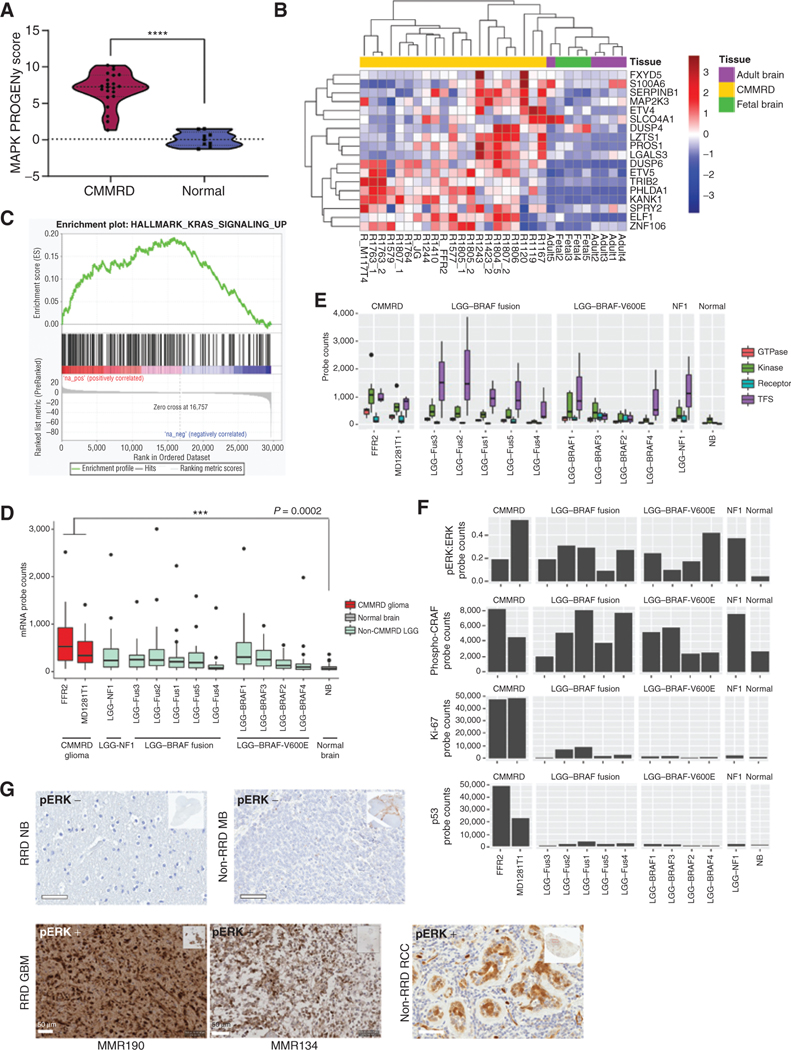
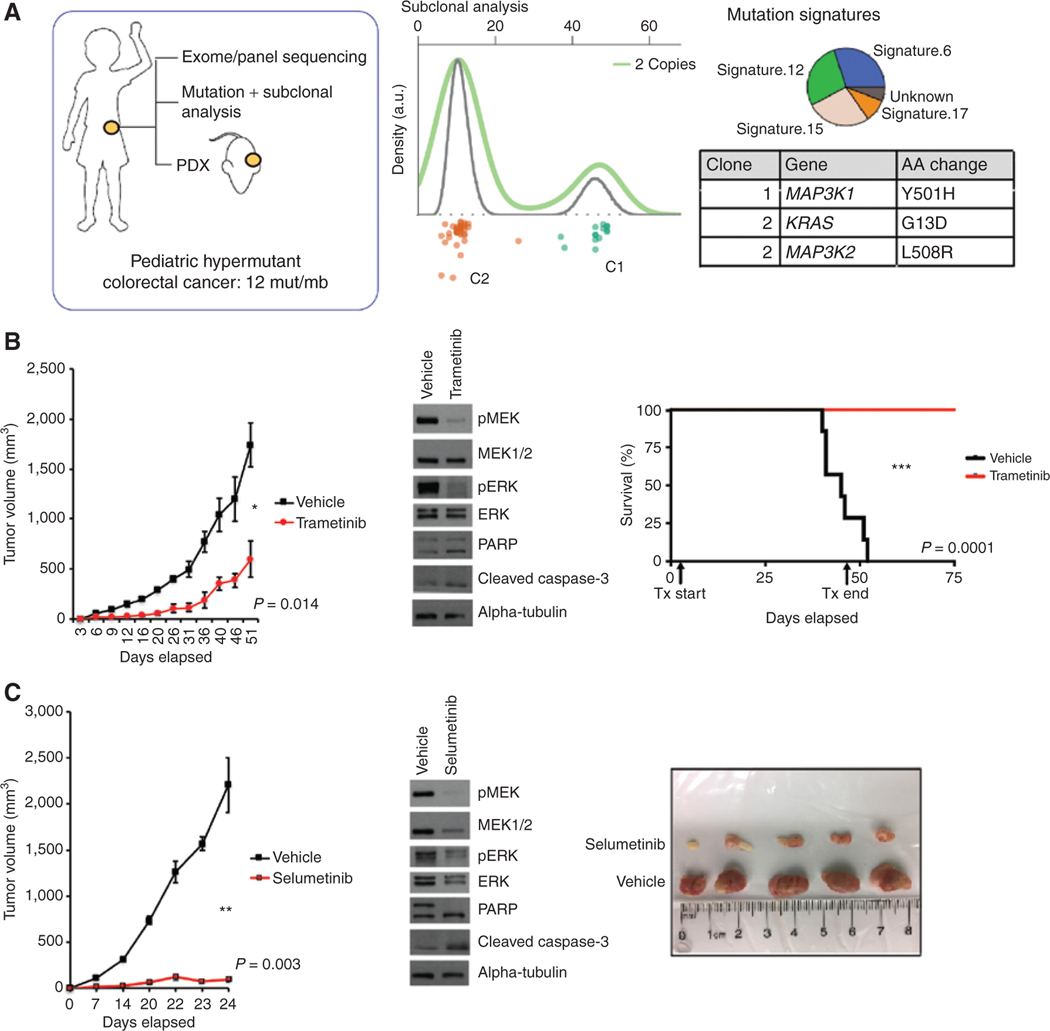
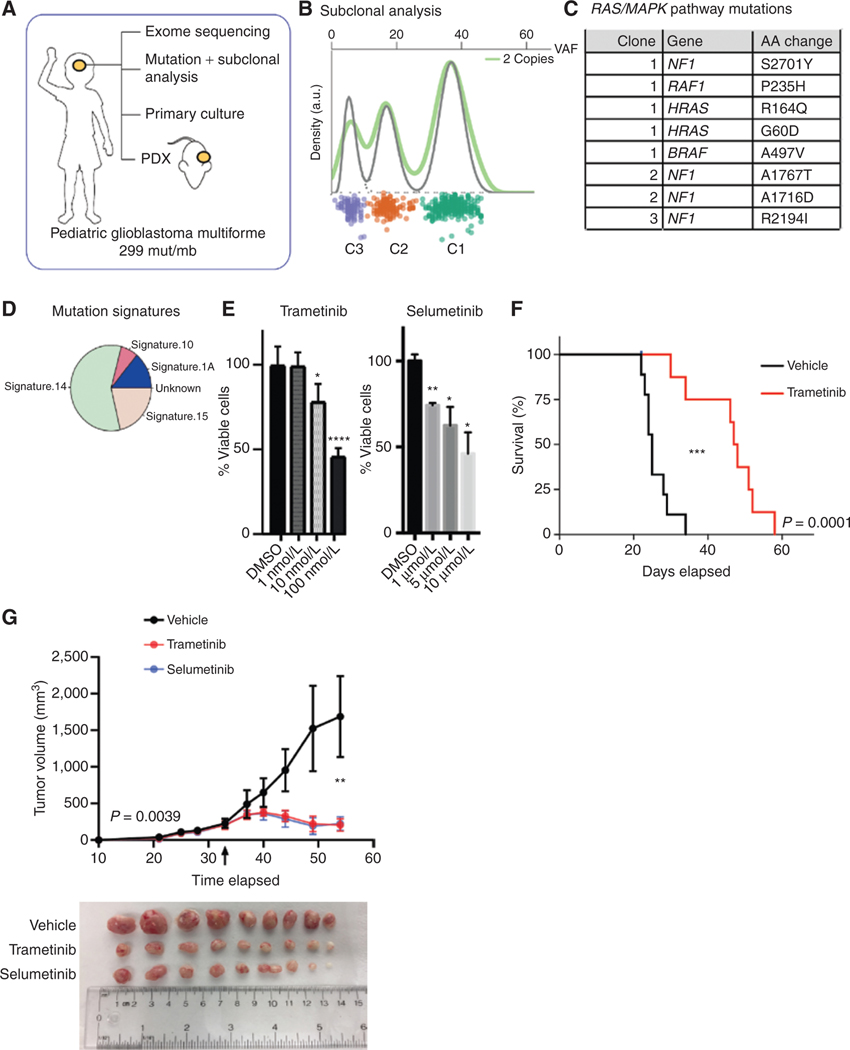
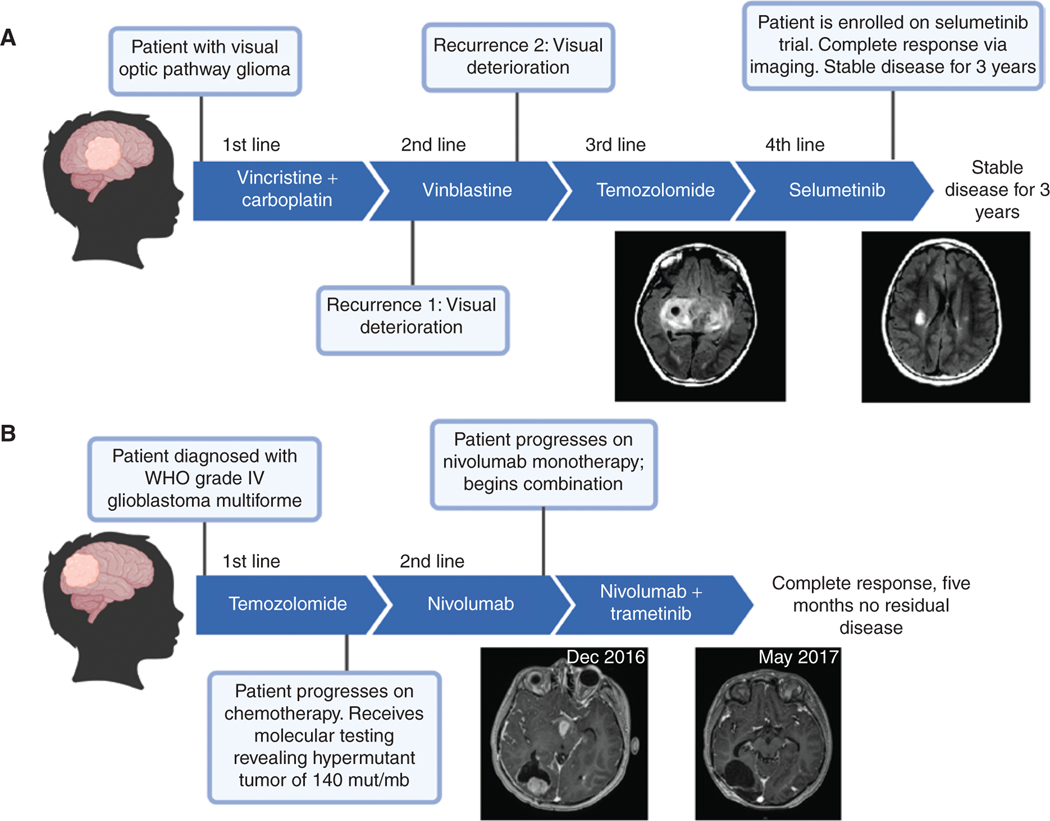
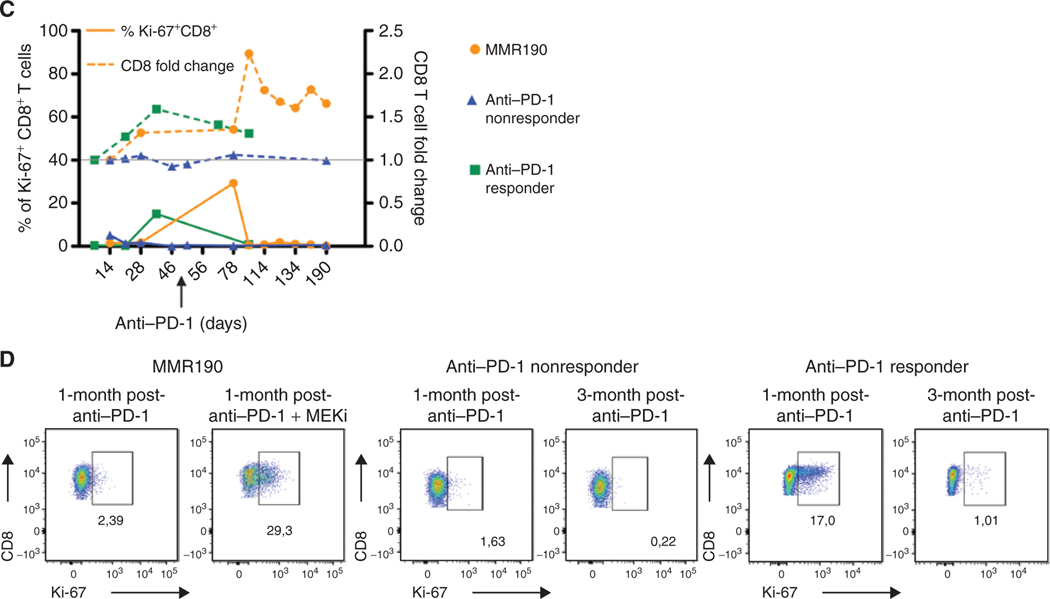
The RAS/MAPK pathway is an emerging targeted pathway across a spectrum of both adult and pediatric cancers. Typically, this is associated with a single, well-characterized point mutation in an oncogene. Hypermutant tumors that harbor many somatic mutations may obscure the interpretation of such targetable genomic events. We find that replication repair-deficient (RRD) cancers, which are universally hypermutant and affect children born with RRD cancer predisposition, are enriched for RAS/MAPK mutations (P = 10-8). These mutations are not random, exist in subclones, and increase in allelic frequency over time. The RAS/MAPK pathway is activated both transcriptionally and at the protein level in patient-derived RRD tumors, and these tumors responded to MEK inhibition in vitro and in vivo. Treatment of patients with RAS/MAPK hypermutant gliomas reveals durable responses to MEK inhibition. Our observations suggest that hypermutant tumors may be addicted to oncogenic pathways, resulting in favorable response to targeted therapies. SIGNIFICANCE: Tumors harboring a single RAS/MAPK driver mutation are targeted individually for therapeutic purposes. We find that in RRD hypermutant cancers, mutations in the RAS/MAPK pathway are enriched, highly expressed, and result in sensitivity to MEK inhibitors. Targeting an oncogenic pathway may provide therapeutic options for these hypermutant polyclonal cancers.This article is highlighted in the In This Issue feature, p. 1307.
©2021 American Association for Cancer Research.
Conflict of interest statement
Authors’ Disclosures
No disclosures were reported by the other authors.
Figures
References
-
- Gargiulo P, Della Pepa C, Berardi S, Califano D, Scala S, Buonaguro L, et al. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated endometrial cancers: new candidates for checkpoint blockade immunotherapy? Cancer Treat Rev 2016;48:61–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
