

Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma
- PMID: 33568347
- PMCID: PMC8895627
- DOI: 10.1158/1078-0432.CCR-20-3363
Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma
Erratum in
-
Correction: Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma.Clin Cancer Res. 2021 Aug 15;27(16):4664. doi: 10.1158/1078-0432.CCR-21-2433. Clin Cancer Res. 2021. PMID: 34389658 No abstract available.
Abstract
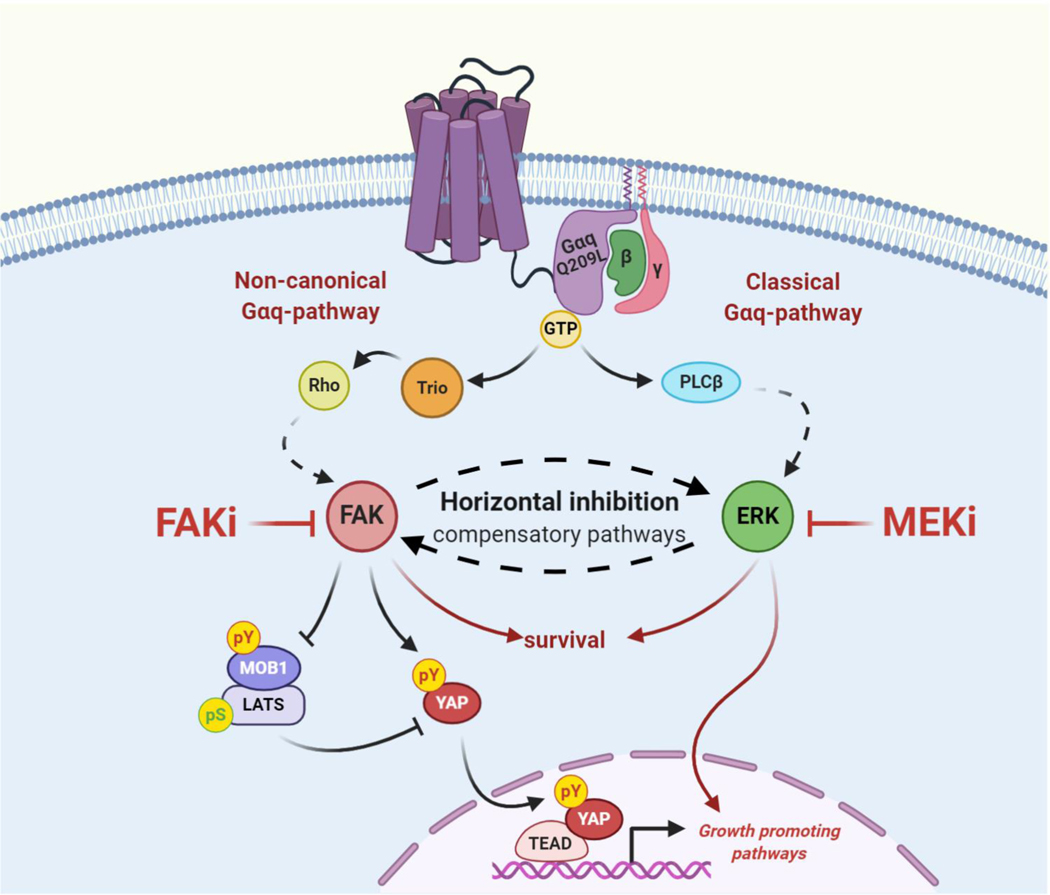
Purpose: Uveal melanoma is the most common eye cancer in adults. Approximately 50% of patients with uveal melanoma develop metastatic uveal melanoma (mUM) in the liver, even after successful treatment of the primary lesions. mUM is refractory to current chemo- and immune-therapies, and most mUM patients die within a year. Uveal melanoma is characterized by gain-of-function mutations in GNAQ/GNA11, encoding Gαq proteins. We have recently shown that the Gαq-oncogenic signaling circuitry involves a noncanonical pathway distinct from the classical activation of PLCβ and MEK-ERK. GNAQ promotes the activation of YAP1, a key oncogenic driver, through focal adhesion kinase (FAK), thereby identifying FAK as a druggable signaling hub downstream from GNAQ. However, targeted therapies often activate compensatory resistance mechanisms leading to cancer relapse and treatment failure.
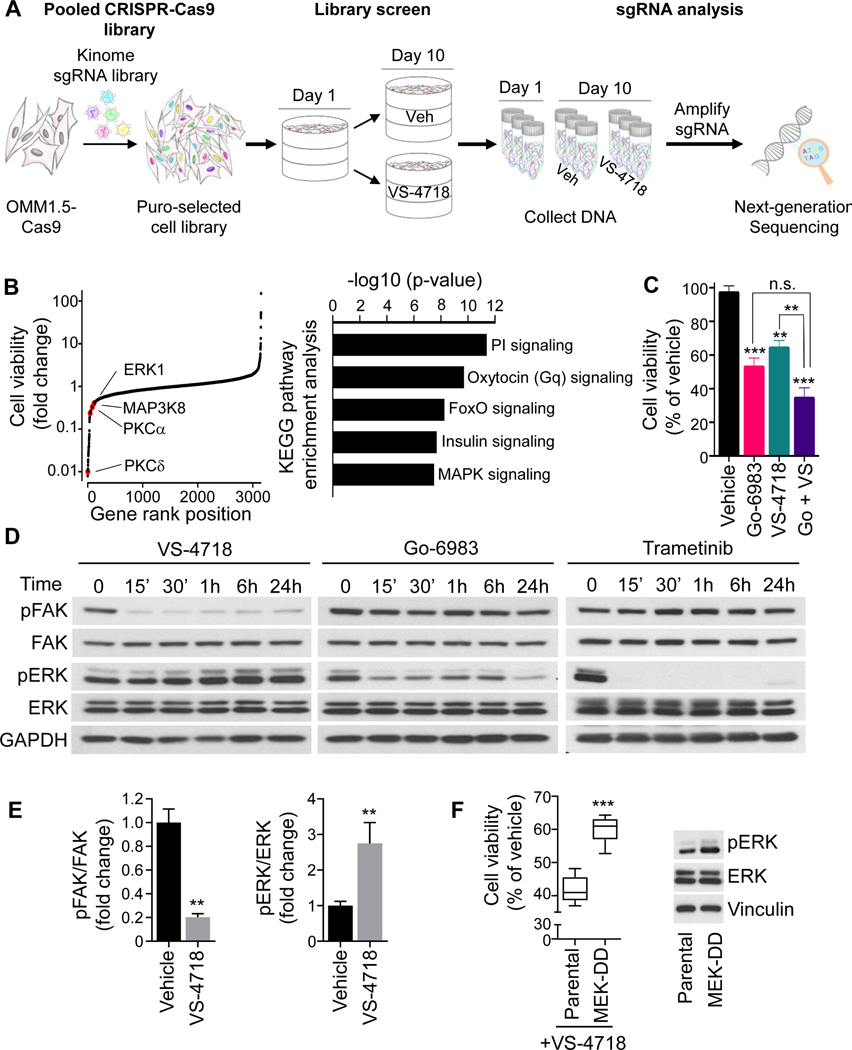
Experimental design: We performed a kinome-wide CRISPR-Cas9 sgRNA screen to identify synthetic lethal gene interactions that can be exploited therapeutically. Candidate adaptive resistance mechanisms were investigated by cotargeting strategies in uveal melanoma and mUM in vitro and in vivo experimental systems.
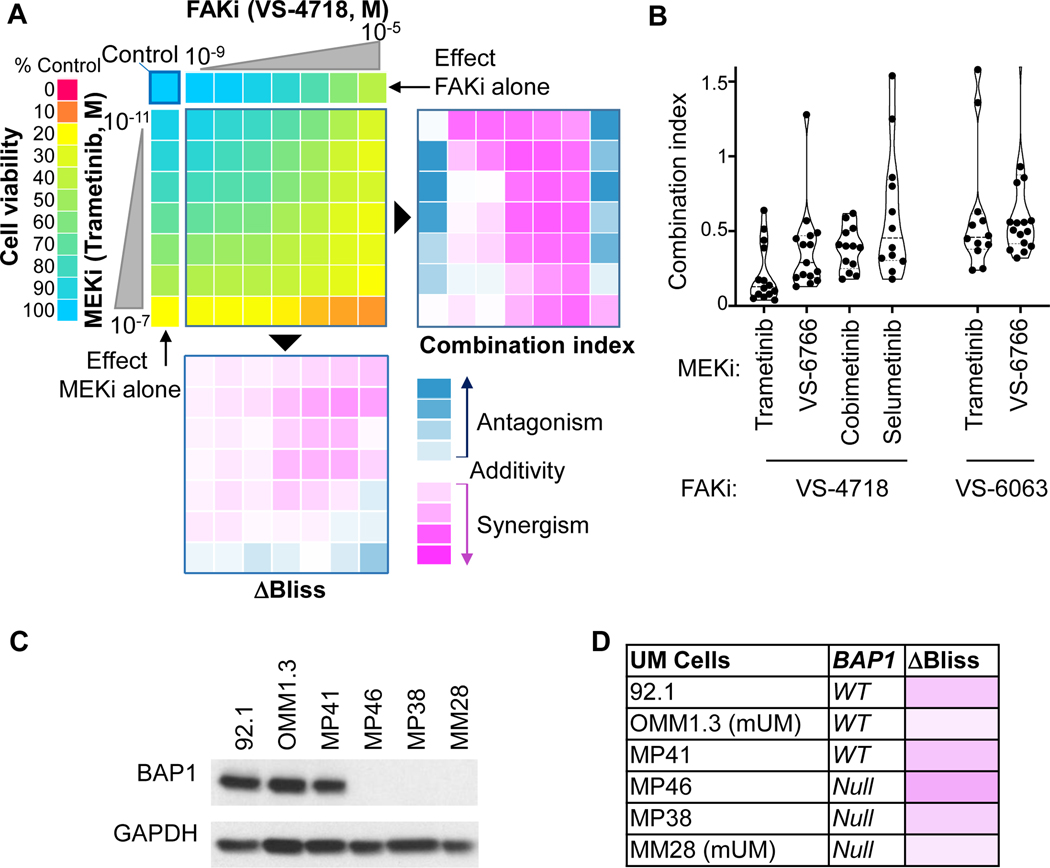
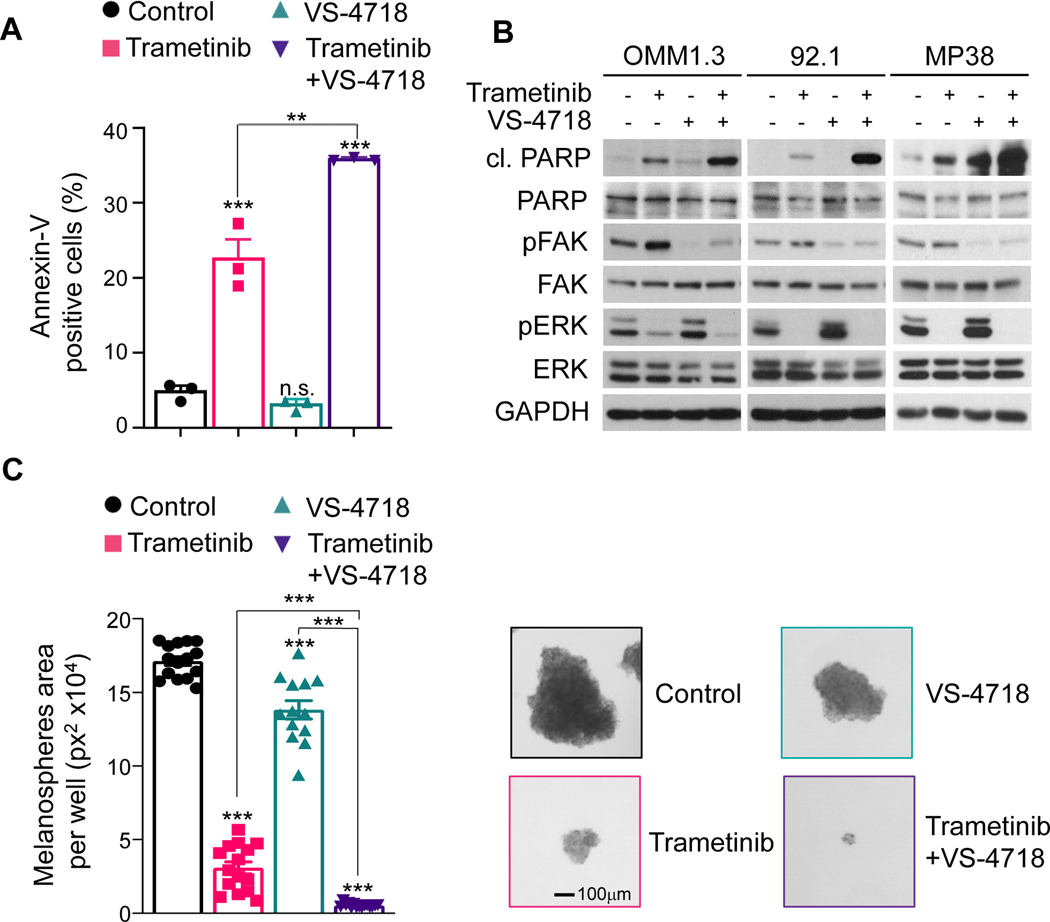
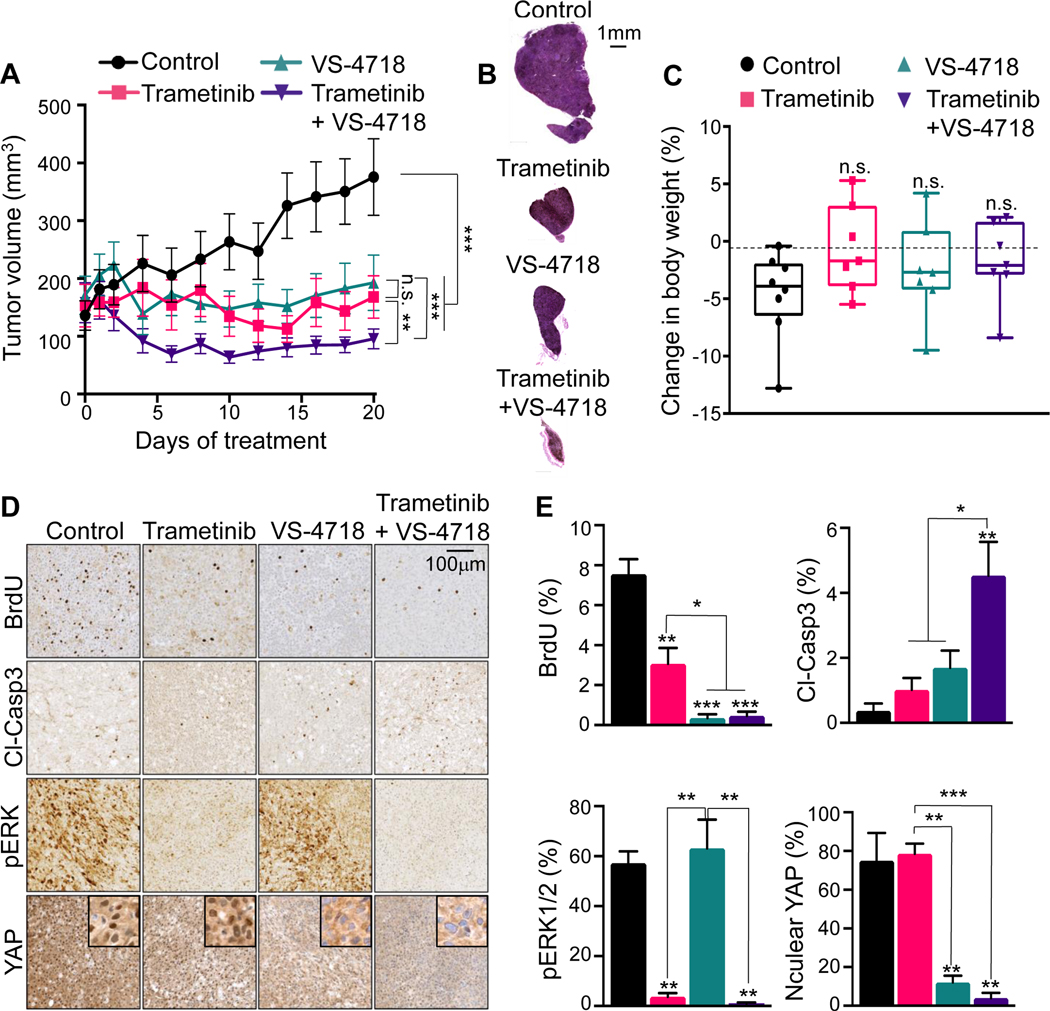
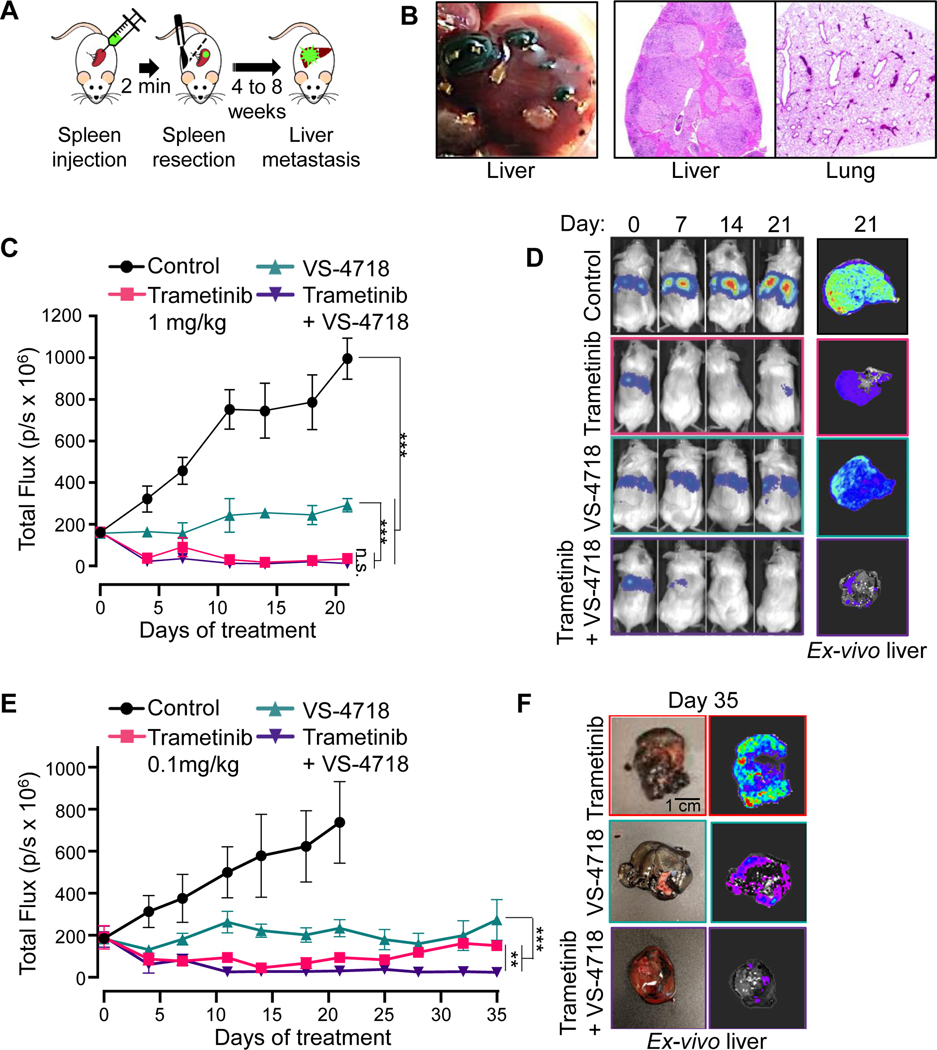
Results: sgRNAs targeting the PKC and MEK-ERK signaling pathways were significantly depleted after FAK inhibition, with ERK activation representing a predominant resistance mechanism. Pharmacologic inhibition of MEK and FAK showed remarkable synergistic growth-inhibitory effects in uveal melanoma cells and exerted cytotoxic effects, leading to tumor collapse in uveal melanoma xenograft and liver mUM models in vivo.
Conclusions: Coupling the unique genetic landscape of uveal melanoma with the power of unbiased genetic screens, our studies reveal that FAK and MEK-ERK cotargeting may provide a new network-based precision therapeutic strategy for mUM treatment.See related commentary by Harbour, p. 2967.
©2021 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
Therapeutic Escape in Gαq-mutant Uveal Melanoma: It's a FAK.Clin Cancer Res. 2021 Jun 1;27(11):2967-2969. doi: 10.1158/1078-0432.CCR-21-0567. Epub 2021 Mar 25. Clin Cancer Res. 2021. PMID: 33766812 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
